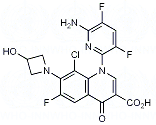
Delafloxacin ist ein Fluorchinolon, es hemmt die bakterielle Topoisomerase IV und DNS-Gyrase (Topoisomerase II). Diese Enzyme werden für die bakterielle Replikation, Transkription, Reparatur und Rekombination der DNS benötigt. Es wirkt gegen grampositive und gramnegative Mikroorganismen, z. b. Staphylokokken, Streptokokken, Enterokokken, E. coli, Enterobacter, Klebsiellen, Proteus mirabilis und Pseudomonas aeruginosa.
Quelle
EPAR der EMA