Der Hersteller informiert in einem Rote-Hand-Brief über wichtige Sicherheitsaspekte und über die Einführung eines Schwangerschaftsverhütungsprogramms.
Vismodegib wirkt teratogen. Es kann zu embryofetalem Tod oder schweren Geburtsfehlern führen und darf während der Schwangerschaft nicht angewendet werden.
Vismodegib darf nur von einem in der Behandlung der zugelassenen Indikation erfahrenen Arzt verschrieben werden, der die Patienten über alle Sicherheitsmaßnahmen bezüglich der Anwendung aufklären muss. Vismodegib darf während der Schwangerschaft nicht angewendet werden: Frauen im gebärfähigen Alter und Männer (Vismodegib kann in Spermien enthalten sein) müssen während und nach der Behandlung Maßnahmen zur Schwangerschaftsverhütung einhalten. Hierzu wurde ein Schwangerschaftsverhütungsprogramm entwickelt. Während der Behandlung mit Vismodegib und noch 24 Monate danach darf nicht gestillt und kein Blut gespendet werden.
Quelle:
AkdÄ Drug Safety Mail vom 31. Juli 2013
Mittwoch, 31. Juli 2013
Pazopanib: Überwachung der Hepatotoxizität
Der Hersteller von Pazopanib (Votrient) informiert über Änderungen zur Häufigkeit von Leberfunktionsuntersuchungen.
Untersuchungen sollten vor Beginn der Behandlung und danach in den Wochen 3, 5, 7 und 9 durchgeführt werden. Danach Untersuchungen in den Monaten 3 und 4 und anschließend in regelmäßigen Abständen wie klinisch indiziert. Bei erhöhten Leberfunktionswerten sollten die Kontrollen häufiger durchgeführt oder Pazopanib vorübergehend beziehungsweise endgültig abgesetzt werden.
Quellen:
Informationsbrief und neue Fachinformation vom Juli 2013
AkdÄ Drug Safety Mail vom 30. Juli 2013
Untersuchungen sollten vor Beginn der Behandlung und danach in den Wochen 3, 5, 7 und 9 durchgeführt werden. Danach Untersuchungen in den Monaten 3 und 4 und anschließend in regelmäßigen Abständen wie klinisch indiziert. Bei erhöhten Leberfunktionswerten sollten die Kontrollen häufiger durchgeführt oder Pazopanib vorübergehend beziehungsweise endgültig abgesetzt werden.
Quellen:
Informationsbrief und neue Fachinformation vom Juli 2013
AkdÄ Drug Safety Mail vom 30. Juli 2013
Metoclopramid: EMA empfiehlt Einschränkung der Zulassung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Metoclopramid einzuschränken.
Aufgrund einer Sicherheitsprüfung von Metoclopramid, welche die französische Arzneimittelagentur ANSM veranlasst hatte, kommt der CHMP zu dem Ergebnis, dass der Einsatz von Metoclopramid in vielen Indikationen aufgrund eines negativen Nutzen-Risikoverhältnisses nicht mehr zu rechtfertigen ist.
Um das Risiko von tardiven Dyskinesien zu senken, soll Metoclopramid künftig nur noch über maximal 5 Tage eingesetzt werden. Damit entfallen Indikationen wie Gastroparese, Dyspepsie oder gastroösophageale Refluxkrankheit, da es sich meist um chronische Erkrankungen handelt, bei denen Metoclopramid bisher über einen längeren Zeitraum eingesetzt wurde.
Nicht betroffen ist der Einsatz von Metoclopramid zur Prävention von Übelkeit und Erbrechen nach Operationen (PONV), nach Radiotherapie sowie nach Chemotherapie (hier nur bei verzögerter, nicht aber akuter Nausea und Vomitus). Auch zur symptomatischen Therapie von Übelkeit und Erbrechen, einschließlich der akuten Migräne darf Metoclopramid weiter eingesetzt werden
Die maximale Tagedosis soll bei Kindern und Erwachsenen auf 0,5 mg pro Kilogramm Körpergewicht beschränkt werden. Die Einzeldosis bei Erwachsenen soll 10 mg nicht überschreiten, bei Kindern sollten es nicht mehr als 0,1 bis 0,15 mg Metoclopramid pro Kilogramm Körpergewicht sein. Die Einzeldosis soll nicht häufiger als dreimal am Tag eingenommen werden.
Da die Einnahme von flüssigen Formulierungen bei Kindern in der Vergangenheit immer wieder zu Überdosierungen geführt hat, soll die Dosisstärke auf maximal 1 mg/ml beschränkt werden. Intravenöse Formulierungen mit Konzentrationen über 5 mg/ml sollen ebenfalls verschwinden, ebenso Suppositorien mit mehr als 20 mg. Die intravenöse Gabe soll künftig langsam über einen Zeitraum von mindestens 3 Minuten erfolgen, um die Gefahr von Nebenwirkungen zu minimieren.
Quelle:
Pressemitteilung der EMA vom 26. Juli 2013
Aufgrund einer Sicherheitsprüfung von Metoclopramid, welche die französische Arzneimittelagentur ANSM veranlasst hatte, kommt der CHMP zu dem Ergebnis, dass der Einsatz von Metoclopramid in vielen Indikationen aufgrund eines negativen Nutzen-Risikoverhältnisses nicht mehr zu rechtfertigen ist.
Um das Risiko von tardiven Dyskinesien zu senken, soll Metoclopramid künftig nur noch über maximal 5 Tage eingesetzt werden. Damit entfallen Indikationen wie Gastroparese, Dyspepsie oder gastroösophageale Refluxkrankheit, da es sich meist um chronische Erkrankungen handelt, bei denen Metoclopramid bisher über einen längeren Zeitraum eingesetzt wurde.
Nicht betroffen ist der Einsatz von Metoclopramid zur Prävention von Übelkeit und Erbrechen nach Operationen (PONV), nach Radiotherapie sowie nach Chemotherapie (hier nur bei verzögerter, nicht aber akuter Nausea und Vomitus). Auch zur symptomatischen Therapie von Übelkeit und Erbrechen, einschließlich der akuten Migräne darf Metoclopramid weiter eingesetzt werden
Die maximale Tagedosis soll bei Kindern und Erwachsenen auf 0,5 mg pro Kilogramm Körpergewicht beschränkt werden. Die Einzeldosis bei Erwachsenen soll 10 mg nicht überschreiten, bei Kindern sollten es nicht mehr als 0,1 bis 0,15 mg Metoclopramid pro Kilogramm Körpergewicht sein. Die Einzeldosis soll nicht häufiger als dreimal am Tag eingenommen werden.
Da die Einnahme von flüssigen Formulierungen bei Kindern in der Vergangenheit immer wieder zu Überdosierungen geführt hat, soll die Dosisstärke auf maximal 1 mg/ml beschränkt werden. Intravenöse Formulierungen mit Konzentrationen über 5 mg/ml sollen ebenfalls verschwinden, ebenso Suppositorien mit mehr als 20 mg. Die intravenöse Gabe soll künftig langsam über einen Zeitraum von mindestens 3 Minuten erfolgen, um die Gefahr von Nebenwirkungen zu minimieren.
Quelle:
Pressemitteilung der EMA vom 26. Juli 2013
Ketoconazol: EMA empfiehlt Marktrücknahme
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung für orale Ketoconazol-Zubereitungen zu widerrufen.
Die orale Formulierung von Ketoconazol soll aufgrund der nachgewiesenen Hepatotoxizität verboten werden. Die topischen Präparate sind nicht von den Einschränkungen betroffen.
Die Überprüfung des Nutzen-Risiko-Verhältnisses ging von der französischen Behörde aus. Sie stufte angesichts neuerer besser verträglicher Mittel wie Fluconazol und Itraconazol - die allerdings auch nicht frei von hepatotoxischen Nebenwirkungen sind – das Nutzen-Risiko-Verhältnis von Ketoconazol als negativ eingestuft. Dies wird nun vom Ausschuss für Humanarzneimittel der EMA bestätigt. Den nationalen Behörden wird allerdings freigestellt, eine Verwendung zur Behandlung des Cushing-Syndroms zu erlauben.
Quelle:
Pressemitteilung der EMA vom 25. Juli 2013
Die orale Formulierung von Ketoconazol soll aufgrund der nachgewiesenen Hepatotoxizität verboten werden. Die topischen Präparate sind nicht von den Einschränkungen betroffen.
Die Überprüfung des Nutzen-Risiko-Verhältnisses ging von der französischen Behörde aus. Sie stufte angesichts neuerer besser verträglicher Mittel wie Fluconazol und Itraconazol - die allerdings auch nicht frei von hepatotoxischen Nebenwirkungen sind – das Nutzen-Risiko-Verhältnis von Ketoconazol als negativ eingestuft. Dies wird nun vom Ausschuss für Humanarzneimittel der EMA bestätigt. Den nationalen Behörden wird allerdings freigestellt, eine Verwendung zur Behandlung des Cushing-Syndroms zu erlauben.
Quelle:
Pressemitteilung der EMA vom 25. Juli 2013
Zonisamid: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Zonisamid (Zonegran, Eisai) auf die Behandlung von Jugendlichen und Kindern ab sechs Jahren mit partiellen epileptischen Anfällen zu erweitern.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Ustekinumab: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Ustekinumab (Stelara, Janssen-Cilag) zu erweitern. Es soll nun auch in Kombination mit Methotrexat zur Behandlung von Erwachsenen mit Psoriasis-Arthritis eingesetzt werden können, wenn diese nicht ausreichend auf DMARDs angesprochen haben.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Golimumab: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Golimumab (Simponi, Janssen) auf die Behandlung der Colitis ulcerosa zu erweitern, wenn die Patienten auf eine konventionelle Therapie nicht ausreichend ansprechen oder wenn Kontraindikationen bestehen.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Eltrombopag: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Eltrombopag (Revolade, GSK) zu erweitern. Es soll nun auch bei Erwachsenen mit Hepatitis-C-Virus-Infektion für die Behandlung der Thrombozytopenie eingesetzt werden können, wenn sie eine optimale Interferon-basierte Therapie einschränkt oder verhindert.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Darunavir: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Darunavir (Prezista, Janssen-Cilag) zu erweitern. Es soll nun auch für die Behandlung von Kindern mit HIV-Infektion ab einem Alter von drei Jahren und einem Körpergewicht von mindestens 15 kg eingesetzt werden können.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Dienstag, 30. Juli 2013
Canakinumab: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Indikation von Canakinumab (Ilaris, Novartis) zu erweitern, es soll nun auch bei Kindern ab zwei Jahren mit aktiver systemischer juveniler Arthritis eingesetzt werden können, die nicht ausreichend auf NSAR und systemische Corticoide angesprochen haben.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Aflibercept: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, die Zulassung von Aflibercept (Eylea, Bayer) zu erweitern, es soll nun auch zur Behandlung von Sehstörungen bei Erwachsenen aufgrund eines Makulaödems wegen eines Zentralvenenverschlusses eingesetzt werden können.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Samstag, 27. Juli 2013
Defibrotid: EMA empfiehlt Zulassung bei hepatischer venookklusiver Erkrankung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, Defibrotid (Defitelio, Gentium) für die Behandlung von Patienten mit schwerer hepatischer venookklusiver Erkrankung zuzulassen.
Defibrotid st ein Derivat von aus Schweinedarm isolierter DNA, das protektive Effekte auf vaskuläre Endothelzellen besonders bei den kleinen Gefäßen hat. Es wirkt antithrombotisch, antientzündlich und antiischämisch. Es soll bei hepatischer veno-okklusiver Erkrankung (VOD), die nach autologer und allogener Stammzelltransplantation auftreten kann, zugelassen werden.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Defibrotid st ein Derivat von aus Schweinedarm isolierter DNA, das protektive Effekte auf vaskuläre Endothelzellen besonders bei den kleinen Gefäßen hat. Es wirkt antithrombotisch, antientzündlich und antiischämisch. Es soll bei hepatischer veno-okklusiver Erkrankung (VOD), die nach autologer und allogener Stammzelltransplantation auftreten kann, zugelassen werden.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Delamanid: EMA empfiehlt Zulassung bei Tuberkulose nicht
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, Delamanid (Delamanid, Otsuka) nicht für die Behandlung von Patienten mit multiresistenter Tuberkulose zuzulassen, weil der Nutzen bislang nicht ausreichend belegt ist.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Cobicistat: EMA empfiehlt Zulassung als Pharmakoenhancer
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, Cobicistat (Tybost, Gilead) als pharmakokinetischen Enhancer für die Proteaseinhibitoren Atazanavir und Darunavir zuzulassen. Cobicistat ist in der Vierfachkombination Stribild bereits im Handel verfügbar.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Alogliptin: EMA empfiehlt Zulassung für Diabetes mellitus Typ 2
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, Alogliptin (Vipidia, Takeda) allein sowie in Kombination mit Metformin (Vipdomet, Takeda) und Pioglitazon (Incresync, Takeda) für die Behandllung von Patienten mit Diabetes mellitus zuzulassen.
Alogliptin (Vipidia, Takeda) ist wie z. B. Sitagliptin ein DPP-IV-Hemmer. Er soll für die Behandlung von Patienten ab 18 Jahren mit Diabetes mellitus Typ 2 zugelassen werden, um in Kombination mit anderen Bllutglucose-senkenden Maßnahmen die glykämische Kontrolle zu verbessern, wenn diese bislang nicht ausreichend wirksam waren.

Quelle:
Mitteilung der EMA vom 25. Juli 2013
Alogliptin (Vipidia, Takeda) ist wie z. B. Sitagliptin ein DPP-IV-Hemmer. Er soll für die Behandlung von Patienten ab 18 Jahren mit Diabetes mellitus Typ 2 zugelassen werden, um in Kombination mit anderen Bllutglucose-senkenden Maßnahmen die glykämische Kontrolle zu verbessern, wenn diese bislang nicht ausreichend wirksam waren.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Afatinib: EMA empfiehlt Zulassung für NSCLC
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juli 2013 empfohlen, Afatinib (Giotrif, Boehringer Ingelheim) für die Behandlung von Patienten mit NSCLC und aktivierenden EGFR-Mutationen zuzulassen.
Afatinib (Giotrif, Boehringer Ingelheim) ist ein Proteinkinasehemmer, der irreversibel alle Signale von Homo- und Heterodimeren der ErB-Familienmitglieder wie EGFR, Her2, ErB3 und ErB4 hemmt.
 Er soll als Monotherapie zur Behandlung von EGFR-TKI-naiven-Patienten mit lokal fortgeschrittenem oder metastasiertem nicht kleinzelligen Lungenkarzinom (NSCLC) mit aktivierenden EGFR-Mutationen zugelassen werden. Als Teil der Zulassung muss ein Pharmakovigilanzprogramm implementiert werden.
Er soll als Monotherapie zur Behandlung von EGFR-TKI-naiven-Patienten mit lokal fortgeschrittenem oder metastasiertem nicht kleinzelligen Lungenkarzinom (NSCLC) mit aktivierenden EGFR-Mutationen zugelassen werden. Als Teil der Zulassung muss ein Pharmakovigilanzprogramm implementiert werden.
Der Nutzen von Afatinib wurde in einer randomisierten offenen Phase-III-Studie im Vergleich zu Chemotherapie nachgewiesen. Die Behandlung mit Afatinib verlängerte das PFS im Vergleich zur Chemotherapie. Häufigste Nebenwirkungen waren Diarrhö, Stomatitis, Hautausschlag, akneiforme Dermatitis, Juckreiz, trockene Haut, Paronychie, vermindertes Appetit und Nasenbluten.
Die FDA hat Afatinib für diese Indikation im Juli 2013 zugelassen.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Afatinib (Giotrif, Boehringer Ingelheim) ist ein Proteinkinasehemmer, der irreversibel alle Signale von Homo- und Heterodimeren der ErB-Familienmitglieder wie EGFR, Her2, ErB3 und ErB4 hemmt.

Der Nutzen von Afatinib wurde in einer randomisierten offenen Phase-III-Studie im Vergleich zu Chemotherapie nachgewiesen. Die Behandlung mit Afatinib verlängerte das PFS im Vergleich zur Chemotherapie. Häufigste Nebenwirkungen waren Diarrhö, Stomatitis, Hautausschlag, akneiforme Dermatitis, Juckreiz, trockene Haut, Paronychie, vermindertes Appetit und Nasenbluten.
Die FDA hat Afatinib für diese Indikation im Juli 2013 zugelassen.
Quelle:
Mitteilung der EMA vom 25. Juli 2013
Freitag, 19. Juli 2013
Avanafil von der EU-Kommission zugelassen
Die EU-Kommission hat Avanafil (Spedra, Vivus) für die Behandlung der erektilen Dysfunktion bei erwachsenen Männern zugelassen.
Avanafil (Spedra, Vivus) ist wie Tadalafil, Vardenafil und Sildenafil ein Typ-5-Phosphodiesterasehemmer (PDE 5), es hemmt selektiv und reversibel die zyklo-Guanosinmonophosphat(cGMP)-spezifische Phosphodiesterase 5. Sobald die sexuelle Stimulation zu einer lokalen Freisetzung von Stickstoffmonoxid (NO) führt, bewirkt Avanafil eine Hemmung der PDE5 und damit eine erhöhte Konzentration von cGMP im Schwellkörper des Penis. Dies führt zu einer Entspannung der glatten Muskulatur und einem Bluteinstrom in das Penisgewebe, wodurch es zur Erektion kommt. Avanafil hat bei fehlender sexueller Stimulation keine Wirkung.

Quelle:
Epar der EMA
Avanafil (Spedra, Vivus) ist wie Tadalafil, Vardenafil und Sildenafil ein Typ-5-Phosphodiesterasehemmer (PDE 5), es hemmt selektiv und reversibel die zyklo-Guanosinmonophosphat(cGMP)-spezifische Phosphodiesterase 5. Sobald die sexuelle Stimulation zu einer lokalen Freisetzung von Stickstoffmonoxid (NO) führt, bewirkt Avanafil eine Hemmung der PDE5 und damit eine erhöhte Konzentration von cGMP im Schwellkörper des Penis. Dies führt zu einer Entspannung der glatten Muskulatur und einem Bluteinstrom in das Penisgewebe, wodurch es zur Erektion kommt. Avanafil hat bei fehlender sexueller Stimulation keine Wirkung.
Quelle:
Epar der EMA
Knorpelzellzubereitung von der EU-Kommission zugelassen
Die EU-Kommission hat MACI 500.000 bis 1.000.000 Zellen/cm2 Matrix zur Implantation zugelassen.
MACI ist indiziert für die Wiederherstellung symptomatischer Vollschicht-Knorpeldefekte des Knies
(Grad 3 und 4 auf der modifizierten Outerbridge-Skala) mit einer Größe von 3-20 cm2
bei erwachsenen Patienten mit geschlossener Wachstumsfuge. Es handelt sich um charakterisierte, vitale, autologe, ex vivo expandierte Chondrozyten, die chondrozytenspezifische Markergene exprimieren und auf eine aus Schweinen gewonnene,CE-zertifizierteTyp-I/IIIKollagenmembran aufgebracht wurden. Jede Matrix zur Implantation besteht aus charakterisierten, autologen Chondrozyten in einer Dichte von 500.000 bis 1.000.000 Zellen/cm2 auf einer 14,5 cm² großen Typ-I/III-Kollagenmembran und muss vom Chirurgen entsprechend der Größe und Form des Defekts zugeschnitten werden.
Quelle
EPAR der EMA
MACI ist indiziert für die Wiederherstellung symptomatischer Vollschicht-Knorpeldefekte des Knies
(Grad 3 und 4 auf der modifizierten Outerbridge-Skala) mit einer Größe von 3-20 cm2
bei erwachsenen Patienten mit geschlossener Wachstumsfuge. Es handelt sich um charakterisierte, vitale, autologe, ex vivo expandierte Chondrozyten, die chondrozytenspezifische Markergene exprimieren und auf eine aus Schweinen gewonnene,CE-zertifizierteTyp-I/IIIKollagenmembran aufgebracht wurden. Jede Matrix zur Implantation besteht aus charakterisierten, autologen Chondrozyten in einer Dichte von 500.000 bis 1.000.000 Zellen/cm2 auf einer 14,5 cm² großen Typ-I/III-Kollagenmembran und muss vom Chirurgen entsprechend der Größe und Form des Defekts zugeschnitten werden.
Quelle
EPAR der EMA
Mittwoch, 17. Juli 2013
Flupirtin: Rote-Hand-Brief zur Anwendungseinschränkung
Die Hersteller Flupirtin-haltiger Arzneimittel informieren in einem Rote-Hand-Brief über über das Ergebnis einer neuen Nutzen-Risiko-Bewertung Flupirtin-haltiger Arzneimittel.
Der Pharmakovigilanz-Ausschuss für Risikobewertung (PRAC) der EMA nahm eine Neubewertung der Nutzen und Risiken vor, nachdem Bedenken zur Hepatotoxizität in Verbindung mit Flupirtin-haltigen Arzneimitteln sowie zur Wirksamkeit aufgrund unzureichender Nachweise bei chronischen Schmerzen aufgetreten waren.
Die Beurteilung der Spontanberichte zu Lebererkrankungen unter der Anwendung von Flupirtin, die von einem asymptomatischen Anstieg der Leberenzyme bis zu Leberversagen reichten, hat zu einer Aktualisierung der Fachinformation für Flupirtin-haltige Arzneimittel geführt:
- Flupirtin ist nun für die Behandlung von akuten Schmerzen bei Erwachsenen indiziert und darf nur angewendet werden, wenn eine Behandlung mit anderen Analgetika (z. B. nicht-steroidale Antirheumatika, schwache Opioide) kontraindiziert ist.
- Flupirtin-Lösung zur Injektion (i.m.) ist als Einzeldosis zur Anwendung bei Erwachsenen mit postoperativen Schmerzen indiziert. Ist eine längere Dauer der Anwendung erforderlich, stehen andere Darreichungsformen zur Verfügung. Die Anwendung darf nur erfolgen, wenn eine Behandlung mit anderen Analgetika (z. B. nicht-steroidale Antirheumatika, schwache Opioide) kontraindiziert ist.
- Die Dauer der Behandlung für orale Darreichungsformen und Zäpfchen darf zwei Wochen nicht überschreiten.
- Die Kontraindikationen umfassen nun auch Patienten mit vorbestehenden Lebererkrankungen oder Alkoholmissbrauch, sowie die gleichzeitige Anwendung von Flupirtin mit anderen Medikamenten mit bekannter, klinisch relevanter Hepatotoxizität.
- Leberwertmessungen müssen in wöchentlichen Abständen während der Behandlung durchgeführt werden. Falls abnorme Leberwerte oder klinische Symptome einer Lebererkrankung auftreten, muss die Behandlung abgebrochen werden.
Der Pharmakovigilanz-Ausschuss für Risikobewertung (PRAC) der EMA nahm eine Neubewertung der Nutzen und Risiken vor, nachdem Bedenken zur Hepatotoxizität in Verbindung mit Flupirtin-haltigen Arzneimitteln sowie zur Wirksamkeit aufgrund unzureichender Nachweise bei chronischen Schmerzen aufgetreten waren.
Die Beurteilung der Spontanberichte zu Lebererkrankungen unter der Anwendung von Flupirtin, die von einem asymptomatischen Anstieg der Leberenzyme bis zu Leberversagen reichten, hat zu einer Aktualisierung der Fachinformation für Flupirtin-haltige Arzneimittel geführt:
- Flupirtin ist nun für die Behandlung von akuten Schmerzen bei Erwachsenen indiziert und darf nur angewendet werden, wenn eine Behandlung mit anderen Analgetika (z. B. nicht-steroidale Antirheumatika, schwache Opioide) kontraindiziert ist.
- Flupirtin-Lösung zur Injektion (i.m.) ist als Einzeldosis zur Anwendung bei Erwachsenen mit postoperativen Schmerzen indiziert. Ist eine längere Dauer der Anwendung erforderlich, stehen andere Darreichungsformen zur Verfügung. Die Anwendung darf nur erfolgen, wenn eine Behandlung mit anderen Analgetika (z. B. nicht-steroidale Antirheumatika, schwache Opioide) kontraindiziert ist.
- Die Dauer der Behandlung für orale Darreichungsformen und Zäpfchen darf zwei Wochen nicht überschreiten.
- Die Kontraindikationen umfassen nun auch Patienten mit vorbestehenden Lebererkrankungen oder Alkoholmissbrauch, sowie die gleichzeitige Anwendung von Flupirtin mit anderen Medikamenten mit bekannter, klinisch relevanter Hepatotoxizität.
- Leberwertmessungen müssen in wöchentlichen Abständen während der Behandlung durchgeführt werden. Falls abnorme Leberwerte oder klinische Symptome einer Lebererkrankung auftreten, muss die Behandlung abgebrochen werden.
Quelle:
Montag, 15. Juli 2013
Diclofenac: Rote-Hand-Brief mit neuen KI und Warnhinweisen
In einem
Rote-Hand-Brief vom Juli 2013 informieren die Hersteller von Diclofenac nun über wichtige Einschränkungen bei der Anwendung des NSAR.
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) überprüfte 2012 die vorhandenen Informationen über das Risiko von kardiovaskulären Nebenwirkungen (wie zum Beispiel Herzinfarkt oder Schlaganfall) unter der Therapie mit nichtselektiven NSARs. Der CHMP schlussfolgerte, dass die Daten weitere Beweise für die bisher bekannten Risiken dieser Arzneimittel liefern. Insgesamt wiesen die Studien durchgehend auf ein geringfügig erhöhtes Risiko von kardiovaskulären Nebenwirkungen durch Diclofenac hin, vergleichbar mit dem Risiko unter der Behandlung mit COX-2-Hemmern.
In einem Rote-Hand-Brief vom Juli 2013 informieren die Hersteller nun über wichtige Einschränkungen:
- Die derzeit verfügbaren Daten weisen darauf hin, dass die Therapie mit Diclofenac mit einem erhöhten Risiko arterieller thrombotischer Ereignisse, vergleichbar mit dem von selektiven COX-2-Hemmern, assoziiert ist.
- Diclofenac ist jetzt kontraindiziert bei Patienten mit bestehender Herzinsuffizienz (New York Heart Association, NYHA, Stadien II-IV), ischämischer Herzerkrankung, peripherer Arterienerkrankung oder zerebrovaskulärer Erkrankung. Bei Patienten mit diesen Erkrankungen sollte die Behandlung überprüft werden.
- Die Behandlung mit Diclofenac sollte bei Patienten mit signifikanten Risikofaktoren für kardiovaskuläre Ereignisse (z. B. Hypertonie, Hyperlipidämie, Diabetes mellitus, Rauchen) nur nach sorgfältiger Abwägung begonnen werden.
- Bei allen Patienten sollte die niedrigste wirksame Dosis über den kürzesten, zur Symptomkontrolle erforderlichen Zeitraum angewendet werden.
Quelle:
AkdÄ Drug Safety Mail vom 15. Juli 2013
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) überprüfte 2012 die vorhandenen Informationen über das Risiko von kardiovaskulären Nebenwirkungen (wie zum Beispiel Herzinfarkt oder Schlaganfall) unter der Therapie mit nichtselektiven NSARs. Der CHMP schlussfolgerte, dass die Daten weitere Beweise für die bisher bekannten Risiken dieser Arzneimittel liefern. Insgesamt wiesen die Studien durchgehend auf ein geringfügig erhöhtes Risiko von kardiovaskulären Nebenwirkungen durch Diclofenac hin, vergleichbar mit dem Risiko unter der Behandlung mit COX-2-Hemmern.
In einem Rote-Hand-Brief vom Juli 2013 informieren die Hersteller nun über wichtige Einschränkungen:
- Die derzeit verfügbaren Daten weisen darauf hin, dass die Therapie mit Diclofenac mit einem erhöhten Risiko arterieller thrombotischer Ereignisse, vergleichbar mit dem von selektiven COX-2-Hemmern, assoziiert ist.
- Diclofenac ist jetzt kontraindiziert bei Patienten mit bestehender Herzinsuffizienz (New York Heart Association, NYHA, Stadien II-IV), ischämischer Herzerkrankung, peripherer Arterienerkrankung oder zerebrovaskulärer Erkrankung. Bei Patienten mit diesen Erkrankungen sollte die Behandlung überprüft werden.
- Die Behandlung mit Diclofenac sollte bei Patienten mit signifikanten Risikofaktoren für kardiovaskuläre Ereignisse (z. B. Hypertonie, Hyperlipidämie, Diabetes mellitus, Rauchen) nur nach sorgfältiger Abwägung begonnen werden.
- Bei allen Patienten sollte die niedrigste wirksame Dosis über den kürzesten, zur Symptomkontrolle erforderlichen Zeitraum angewendet werden.
Quelle:
AkdÄ Drug Safety Mail vom 15. Juli 2013
Vismodegib von der EU-Kommission bedingt zugelassen
Die EU-Kommission hat Vismodegib (Erivedge, Roche) für die Behandlung von Patienten mit symptomatischen metastasiertem oder lokal fortgeschrittenem, nicht operablem oder bestrahlbarem Basalzellkarzinom bedingt zugelassen.
Vismodegib (Erivedge, Roche) ist ein oral applizierbares so genannten kleines Molekül, das die überaktive Signalgebung im Hedgehog-Signalweg selektiv hemmt. Dieser gilt als ein zentraler molekularer Mechanismus bei der Entwicklung des Basalzellkarzinoms. Von der FDA war Vismodegib bereits im Januar 2012 zugelassen worden (siehe med|pharm|text-Blog).

Bedingte Zulassungen können erteilt werden, wenn Medikamente eine positive Nutzen-Risiko-Bewertung erhalten, die einen ungedeckten medizinischen Bedarf befriedigen und deren Verfügbarkeit einen erheblichen Nutzen für die öffentliche Gesundheit bringt. Gemäss den Bestimmungen einer bedingten Zulassung wird der Hersteller zusätzliche Daten zu Vismodegib bei fortgeschrittenem Basalzellkarzinom aus einer derzeit laufenden weltweiten Sicherheitsstudie einreichen.
Vismodegib (Erivedge, Roche) ist ein oral applizierbares so genannten kleines Molekül, das die überaktive Signalgebung im Hedgehog-Signalweg selektiv hemmt. Dieser gilt als ein zentraler molekularer Mechanismus bei der Entwicklung des Basalzellkarzinoms. Von der FDA war Vismodegib bereits im Januar 2012 zugelassen worden (siehe med|pharm|text-Blog).
Bedingte Zulassungen können erteilt werden, wenn Medikamente eine positive Nutzen-Risiko-Bewertung erhalten, die einen ungedeckten medizinischen Bedarf befriedigen und deren Verfügbarkeit einen erheblichen Nutzen für die öffentliche Gesundheit bringt. Gemäss den Bestimmungen einer bedingten Zulassung wird der Hersteller zusätzliche Daten zu Vismodegib bei fortgeschrittenem Basalzellkarzinom aus einer derzeit laufenden weltweiten Sicherheitsstudie einreichen.
Samstag, 13. Juli 2013
Afatinib von der FDA bei Lungenkarzinom zugelassen
Die Food and Drug Administration (FDA) hat Afatinib (Gilotrif, Boehringer Ingelheim) für die Behandlung von Patienten mit fortgeschrittenem nicht kleinzelligen Lungenkarzinom zugelassen, die bestimmte EGFR-Mutationen aufweisen.
Afatinib (Gilotrif, Boehringer Ingelheim) ist ein Tyrosinkinase-Inhibitor, der irreversibel Her2- und EGFR-Kinasen hemmt.

Er wird zusammen mit dem dem EGFR RGQ PCR-Test-Kit zugelassen, denn er ist nur bei Patienten mit metastasiertem NSCLC zugelassen, die im EGFR-Gen in Exon 19 Deletionen oder im Exon 21 eine L858R-Substitution aufweisen.
Quelle:
Presseinformation der FDA vom 12. Juli 2013
Afatinib (Gilotrif, Boehringer Ingelheim) ist ein Tyrosinkinase-Inhibitor, der irreversibel Her2- und EGFR-Kinasen hemmt.

Er wird zusammen mit dem dem EGFR RGQ PCR-Test-Kit zugelassen, denn er ist nur bei Patienten mit metastasiertem NSCLC zugelassen, die im EGFR-Gen in Exon 19 Deletionen oder im Exon 21 eine L858R-Substitution aufweisen.
Quelle:
Presseinformation der FDA vom 12. Juli 2013
Montag, 8. Juli 2013
CHMP publiziert Liste von Substanzen, die sich derzeit im Bewertungsverfahren befinden
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat eine Liste der Substanzen veröffentlicht, die sich derzeit im Bewertungsverfahren für eine Zulassung befinden. In der Liste sind der INN und die geplante Indikation aufgeführt: Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use.
Donnerstag, 4. Juli 2013
Ponatinib von der EU-Komission bei CML zugelassen
Die EU-Kommission hat den Proteinkinasehemmer Ponatinib (Iclusig, Ariad) für die Behandlung von Patienten mit CML zugelassen.
Ponatinib (Iclusig, Ariad) ist eine Proteinkinasehemmer, der die BCR-ABL-Kinase hemmt. Er ist nun für die Behandlung von Patienten mit chronisch myeloischer Leukäme und Philadelphia-Chromosom-positiver akuter Lymphoblasten-Leukämie (Ph+-ALL) zugelassen werden, die auf Nilotinib (Tasigna) oder Dasatinib (Sprycel) resistent sind, diese nicht vertragen, bei denen eine erneute Imatinib-Therapie (Glivec) nicht angezeigt ist oder bei denen eine T315I-Mutation vorliegt.
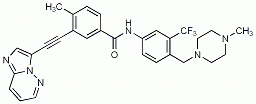
Mit Ponatinib kann bei Patienten mit CML und Ph+-ALL ein zytogenetisches und hämatologisches Ansprechen erreicht werden, auch wenn sie eine T315I-Mutation aufweisen. Häufigste Nebenwirkungen sind Thrombozytenabfall, Hautausschlag, trockene Haut und abdominale Schmerzen.
Ponatinib (Iclusig, Ariad) ist eine Proteinkinasehemmer, der die BCR-ABL-Kinase hemmt. Er ist nun für die Behandlung von Patienten mit chronisch myeloischer Leukäme und Philadelphia-Chromosom-positiver akuter Lymphoblasten-Leukämie (Ph+-ALL) zugelassen werden, die auf Nilotinib (Tasigna) oder Dasatinib (Sprycel) resistent sind, diese nicht vertragen, bei denen eine erneute Imatinib-Therapie (Glivec) nicht angezeigt ist oder bei denen eine T315I-Mutation vorliegt.
Mit Ponatinib kann bei Patienten mit CML und Ph+-ALL ein zytogenetisches und hämatologisches Ansprechen erreicht werden, auch wenn sie eine T315I-Mutation aufweisen. Häufigste Nebenwirkungen sind Thrombozytenabfall, Hautausschlag, trockene Haut und abdominale Schmerzen.
Quelle:
Montag, 1. Juli 2013
Panitumumab: EMA empfiehlt Änderung der Zulassung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, die Zulassung von Panitumumab (Vectibix, Amgen) zu ändern.
Panitumumab soll nun bei Patienten mit metastasiertem Kolorektalkarzinom und RAS-Wildtyp (nicht mehr KRAS-Wildtyp) eingesetzt werden. Als Kontraindikation soll aufgenommen werden, dass die Kombination von Panitumumab und Oxaliplatin-haltiger Chemotherapie nicht bei Patienten mit mutiertem RAS oder unbekannten RAS-Mutationsstatus eingesetzt werden soll.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Panitumumab soll nun bei Patienten mit metastasiertem Kolorektalkarzinom und RAS-Wildtyp (nicht mehr KRAS-Wildtyp) eingesetzt werden. Als Kontraindikation soll aufgenommen werden, dass die Kombination von Panitumumab und Oxaliplatin-haltiger Chemotherapie nicht bei Patienten mit mutiertem RAS oder unbekannten RAS-Mutationsstatus eingesetzt werden soll.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Bortezomib: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, die Zulassung von Bortezomib (Velcade, Janssen-Cilag) zu erweitern.
Bortezomib soll nun in Kombination mit Dexametason oder in Kombination mit Dexamethason und Thalidomid für die Induktionstherapie erwachsener Patienten mit bislang unbehandeltem multiplem Myelom eingesetzt werden können, bei denen eine Hochdosis-Therapie mit hämatopoetischer Stammzelltransplantation möglich ist.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Bortezomib soll nun in Kombination mit Dexametason oder in Kombination mit Dexamethason und Thalidomid für die Induktionstherapie erwachsener Patienten mit bislang unbehandeltem multiplem Myelom eingesetzt werden können, bei denen eine Hochdosis-Therapie mit hämatopoetischer Stammzelltransplantation möglich ist.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Lapatinib: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, die Zulassung von Lapatinib (Tyverb, GSK) zu erweitern, der Tyrosinkinase-Inhibitor soll nun auch in Kombination mit Trastuzumab bei Frauen mit Hormonrezeptor-negativem metastasiertem Mammakarzinom eingesetzt werden können, das unter vorher gehender Trastuzumab- und Chemotherapie progredient war.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Saxagliptin: EMA empfiehlt Zulassungserweiterung zur Monotherapie
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, die Zulassung von Saxagliptin (Onglyza, BMS/AstraZeneca) zu erweitern. Der DPP-IV-Hemmer sol nun auch in Monotherapie bei unzureichend kontrollierten Patienten mit Diabetes mellitus Typ 2 eingesetzt werden können, die Metformin nicht vertragen.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Humanes Fibrinogen/Thrombin: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, die Zulassung des Fibrinklebers Evicel (Omrix) auf den Nahtverschluss beim Verschluss der Dura mater zu erweitern.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Dabrafenib von der EMA zur Zulassung bei Melanom empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, Dabrafenib (Tafinlar, GSK) für die Behandlung von Melanom-Patienten zugzuassen, deren Tumor die BRAF-V600E-Genmutation aufweist.
Dabrafenib ist wie Vemurafenib (Zelboraf) ein oral applizierbarer BRAF-Hemmer. Es soll in Monotherapie für die Behandlung von Erwachsenen mit nicht resezierbarem oder metastasiertem Melanom mit BRAF-V600-Mutation eingesetzt werden.
Als Teil der Zulassung soll ein Pharmakovigilanzprogramm implementiert werden
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Dabrafenib ist wie Vemurafenib (Zelboraf) ein oral applizierbarer BRAF-Hemmer. Es soll in Monotherapie für die Behandlung von Erwachsenen mit nicht resezierbarem oder metastasiertem Melanom mit BRAF-V600-Mutation eingesetzt werden.
Als Teil der Zulassung soll ein Pharmakovigilanzprogramm implementiert werden
Mitteilung der EMA vom 27. Juni 2013
Regorafenib von der EMA zur Zulassung bei Kolorektalkarzinom empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, Regorafenib (Stivarga, Bayer) ist für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zuzulassen.
Regorafenib (Stivarga, Bayer) ist ein Multikinasehemmer, der membrangebundene und intrazelluläre Kinasen hemmt, wie RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, Trk2A, Eph2A, RAF-1, BRAF, BRAFV600E, SAPK2, PTK5 und Abl. Er soll für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zugelassen werden, die zuvor mit einer Fluorpyrimidin/Oxaliplatin- und Irinotecan-basierten Chemotherapie, einer anti-VEGF-Therapie und bei Tumoren vom KRAS-Wildtyp mit einer anti-EGFR-Therapie behandelt worden sind oder für die eine solche Therapie nicht in Frage kommt. Als Teil der Zulassung soll ein Pharmakovigilanzprogramm implementiert werden.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Regorafenib (Stivarga, Bayer) ist ein Multikinasehemmer, der membrangebundene und intrazelluläre Kinasen hemmt, wie RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, Trk2A, Eph2A, RAF-1, BRAF, BRAFV600E, SAPK2, PTK5 und Abl. Er soll für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zugelassen werden, die zuvor mit einer Fluorpyrimidin/Oxaliplatin- und Irinotecan-basierten Chemotherapie, einer anti-VEGF-Therapie und bei Tumoren vom KRAS-Wildtyp mit einer anti-EGFR-Therapie behandelt worden sind oder für die eine solche Therapie nicht in Frage kommt. Als Teil der Zulassung soll ein Pharmakovigilanzprogramm implementiert werden.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Sipuleucel T von der EMA zur Zulassung empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, Sipuleucel T (Provenge, Dendreon) für die Behandlung von Patienten mit Prostatakarzinom zuzulassen.
Sipuleucel (Provenge, Dendreon) ist eine Zubereitung aus autologen peripheren mononukleären Blutzellen, die mit saurer Prostataphospatase (PAP) und GM-CSF aktiviert sind. Diese Immuntherapie soll eine gegen saure Prostataphosphatase (PAP) gerichtete Immunantwort induzieren, ein Antigen, das bei den meisten Prostatakarzinomen exprimiert wird. Sipuleucel T soll für die Behandlung von asymptomatischem oder nur wenig symptomatischen metastasiertem (nicht-viszeralen), kastrationsresistentem Prostatakarzinom bei Männern, bei denen keine Chemotherapie indiziert ist, zugelassen werden.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Sipuleucel (Provenge, Dendreon) ist eine Zubereitung aus autologen peripheren mononukleären Blutzellen, die mit saurer Prostataphospatase (PAP) und GM-CSF aktiviert sind. Diese Immuntherapie soll eine gegen saure Prostataphosphatase (PAP) gerichtete Immunantwort induzieren, ein Antigen, das bei den meisten Prostatakarzinomen exprimiert wird. Sipuleucel T soll für die Behandlung von asymptomatischem oder nur wenig symptomatischen metastasiertem (nicht-viszeralen), kastrationsresistentem Prostatakarzinom bei Männern, bei denen keine Chemotherapie indiziert ist, zugelassen werden.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Mercaptamin von der EMA zur Zulassung bei nephropathischer Zystinose empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom Juni 2013 empfohlen, Mercaptamin (Procysbi, Raptor) für die Behandlung von Patienten mit nephropathischer Zystinose zuzulassen.
Mercaptamin (Cysteamin) (Procysbi, Raptor) wurde in den USA bereits 1995 für die Behandlung der Zystinose zugelassen. Es wandelt Cystin in Cystein und ein Disulfidgemisch um und verzögert die Entstehung eines Nierenversagens. Häufige Nebenwirkungen sind gastrointestinale Störungen wie Übelkeit, Erbrechen und Durchfall, ferner Anorexie, Lethargie und Pyrexie.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Mercaptamin (Cysteamin) (Procysbi, Raptor) wurde in den USA bereits 1995 für die Behandlung der Zystinose zugelassen. Es wandelt Cystin in Cystein und ein Disulfidgemisch um und verzögert die Entstehung eines Nierenversagens. Häufige Nebenwirkungen sind gastrointestinale Störungen wie Übelkeit, Erbrechen und Durchfall, ferner Anorexie, Lethargie und Pyrexie.
Quelle:
Mitteilung der EMA vom 27. Juni 2013
Nepafenac zur Anwendung am Auge in den Markt eingeführt
Nepafenac(Nevanac, Alcon) wurde am 1. Juli 2013 zur Prophylaxe und Behandlung postoperativer Schmerz- und Entzündungszustände bei Kataraktoperationen sowie zur Verminderung des Risikos postoperativer Makulaödeme in Zusammenhang mit Kataraktoperationen bei Diabetikern in den Markt eingeführt.
Nepafenac(Nevanac, Alcon) wurde bereits im Dezember 2007 von der EU-Kommission für die Anwendung am Auge zugelassen. Die Einführung in Deutschland erfolgte zum 1. Juli 2013. Es ist ist ein Amid-Analogon und ein Prodrug von Amfenac.

Es gelangt schnell durch die Hornhaut und wird in den aktiven nicht-steroidalen Entzündungshemmer Amfenac umgewandelt.
Quelle:
EMA-Epar
Nepafenac(Nevanac, Alcon) wurde bereits im Dezember 2007 von der EU-Kommission für die Anwendung am Auge zugelassen. Die Einführung in Deutschland erfolgte zum 1. Juli 2013. Es ist ist ein Amid-Analogon und ein Prodrug von Amfenac.
Es gelangt schnell durch die Hornhaut und wird in den aktiven nicht-steroidalen Entzündungshemmer Amfenac umgewandelt.
Quelle:
EMA-Epar
Abonnieren
Posts (Atom)


