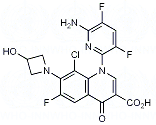
Gilteritinibfumarat ist ein FLT3- und AXL-Inhibitor. Gilteritinib inhibiert die Signalwege des FLT3-Rezeptors und die Proliferation in den Zellen, die FLT3-Mutationen exogen exprimieren, einschließlich FLT3-ITD, FLT3-D835Y und FLT3-ITDD835Y, und es induziert eine Apoptose bei leukämischen Zellen, die FLT3-ITD exprimieren.
Wirksamkeit und Verträglichkeit wurden in der offenen, multizentrischen, randomisierten Phase-3-Studie Admiral untersucht, in der 371 Patienten mit r/r-AMLT und FLT3-Mutation eingeschlossen worden waren. Gilterinib verlängerte im Vergleich zu Chemotherapie das GEsamtüberleben von 5,6 auf 9,3 Monate.
Quelle
EPAR der EMA