Die Food and Drug Administration (FDA) hat die Zulassung von Adalimumab (Humira, Abbott) erweitert, der TNF-alpha-Blocker kann nun auch zur Behandlung von Erwachsenen mit mäßig schwerer bis schwerer Colitis ulcerosa eingesetzt werden.
Quelle:
Pressemitteilung der FDA vom 28. September 2012
Samstag, 29. September 2012
Regorafenib von der FDA für Patienten mit fortgeschrittenem Kolorektalkarzinom zugelassen
Die Food and Drug Administration (FDA) hat Regorafenib (Stivarga, Bayer) für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zugelassen.
Regorafenib (Stivarga, Bayer) ist für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zugelassen, die zuvor mit einer Fluorpyrimidin/Oxaliplatin- und Irinotecan-basierten Chemotherapie, einer anti-VEGF-Therapie und bei Tumoren vom KRAS-Wildtyp mit einer anti-EGFR-Therapie behandelt worden sind. Regorafenib ist ein Multikinasehemmer, der membrangebundene und intrazelluläre Kinasen hemmt, wie RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, Trk2A, Eph2A, RAF-1, BRAF, BRAFV600E, SAPK2, PTK5 und Abl.

Die Zulassung basiert u. a. auf den Ergebnissen einer Phase-III-Studie, mit 760 vorbehandelten Patienten mit metastasiertem Kolorektalkarzinom. In der Regorafenib-Gruppe wurde das Gesamtüberleben signifikant verlängert [Hazard ratio (HR) 0,77 (95% KI: 0,64 - 0,94); p=0,0102]. Die mediane Überlebenszeit betrug 6,4 Monate im Regorafenib-Arm und 5,0 Monate im Plazebo-Arm. Auch das progressionsfreie Überleben war in der Regorafenib-Gruppe signfikant länger [HR 0,49 (95% KI: 0,42-0,58); in den Gesamtansprechraten unterschieden sich die beiden Gruppen nicht.
Schwere Nebenwirkungen in der Regorafenib-Gruppe waren Hepatotoxizität, Blutungen und gastrointestinale Perforationen. Auf das Risiko der hepatoxischen Wirkung muss mit einem Warnhinweis aufmerksam gemacht werden.
Quelle:
Mitteilung der FDA vom 27. September 2012
Regorafenib (Stivarga, Bayer) ist für die Behandlung von Patienten mit metastasiertem Kolorektalkarzinom zugelassen, die zuvor mit einer Fluorpyrimidin/Oxaliplatin- und Irinotecan-basierten Chemotherapie, einer anti-VEGF-Therapie und bei Tumoren vom KRAS-Wildtyp mit einer anti-EGFR-Therapie behandelt worden sind. Regorafenib ist ein Multikinasehemmer, der membrangebundene und intrazelluläre Kinasen hemmt, wie RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, Trk2A, Eph2A, RAF-1, BRAF, BRAFV600E, SAPK2, PTK5 und Abl.

Die Zulassung basiert u. a. auf den Ergebnissen einer Phase-III-Studie, mit 760 vorbehandelten Patienten mit metastasiertem Kolorektalkarzinom. In der Regorafenib-Gruppe wurde das Gesamtüberleben signifikant verlängert [Hazard ratio (HR) 0,77 (95% KI: 0,64 - 0,94); p=0,0102]. Die mediane Überlebenszeit betrug 6,4 Monate im Regorafenib-Arm und 5,0 Monate im Plazebo-Arm. Auch das progressionsfreie Überleben war in der Regorafenib-Gruppe signfikant länger [HR 0,49 (95% KI: 0,42-0,58); in den Gesamtansprechraten unterschieden sich die beiden Gruppen nicht.
Schwere Nebenwirkungen in der Regorafenib-Gruppe waren Hepatotoxizität, Blutungen und gastrointestinale Perforationen. Auf das Risiko der hepatoxischen Wirkung muss mit einem Warnhinweis aufmerksam gemacht werden.
Quelle:
Mitteilung der FDA vom 27. September 2012
Dienstag, 25. September 2012
Rote-Hand-Brief zum Typhus-Polysaccharid-Impfstoff Typhim Vi®
Der Hersteller hat mit einem Rote-Hand-Brief freiwillig bestimmte Chargen des Typhus-Polysaccharid-Impfstoffs Typhim Vi® zurückgerufen, da der Antigengehalt dieser Chargen möglicherweise geringer ist als erwartet.
Darauf deuten die Ergebnisse kürzlich durchgeführter Untersuchungen hin. Der Rückruf betrifft folgende Chargen: G0327-2, G0529-1, G0531-2 und G0531-3. Es bestehen jedoch keine Sicherheitsbedenken für Personen, die mit dem Impfstoff aus einer der zurückgerufenen Chargen geimpft wurden.
Für Personen, die mit Typhim Vi® aus einer der zurückgerufenen Chargen geimpft wurden, wird eine Wiederholungsimpfung zu einem früheren Zeitpunkt als im Normalfall erforderlich nicht empfohlen. Impfstoff aus nicht betroffenen Chargen kann verabreicht werden.
Quelle:
AkdÄ Drug Safety Mail vom 25. September 2012
Darauf deuten die Ergebnisse kürzlich durchgeführter Untersuchungen hin. Der Rückruf betrifft folgende Chargen: G0327-2, G0529-1, G0531-2 und G0531-3. Es bestehen jedoch keine Sicherheitsbedenken für Personen, die mit dem Impfstoff aus einer der zurückgerufenen Chargen geimpft wurden.
Für Personen, die mit Typhim Vi® aus einer der zurückgerufenen Chargen geimpft wurden, wird eine Wiederholungsimpfung zu einem früheren Zeitpunkt als im Normalfall erforderlich nicht empfohlen. Impfstoff aus nicht betroffenen Chargen kann verabreicht werden.
Quelle:
AkdÄ Drug Safety Mail vom 25. September 2012
Catridecacog von der EU-Kommission zur Blutungsprophylaxe zugelassen
Die EU-Kommission hat Catridecacog (Novothirteen, Novo) für die Langzeitprophylaxe von Blutern ab einem Alter von sechs Jahren mit erblich bedingtem Mangen an FXIIIA-Untereinheit zugelassen.
Catridecacog (Novothirteen, Novo) ist eine Untereinheit von Koagulationsfaktor XIII A, die gentechnisch in Hefezellen produziert wird. Sie ist strukturell identisch mit menschlicher F-XIII-A-Unterheinheit [A2], die an die FXIII B-Subunit [A2B2] bindet. Nach Aktivierung verursacht FXIII cross-links von Fibrin und anderen Proteinen, was zu einer verstärkten Resistenz des Gerinnsels gegebenüber der Fibrinolyse führt. Bei Patienten mit erblich bedingtem Mangel an FXIIIA-Untereinheit kann die Substitution mitCatridecacog das Risiko von Blutungen verringern.
Quelle:
EMA-Epar
Catridecacog (Novothirteen, Novo) ist eine Untereinheit von Koagulationsfaktor XIII A, die gentechnisch in Hefezellen produziert wird. Sie ist strukturell identisch mit menschlicher F-XIII-A-Unterheinheit [A2], die an die FXIII B-Subunit [A2B2] bindet. Nach Aktivierung verursacht FXIII cross-links von Fibrin und anderen Proteinen, was zu einer verstärkten Resistenz des Gerinnsels gegebenüber der Fibrinolyse führt. Bei Patienten mit erblich bedingtem Mangel an FXIIIA-Untereinheit kann die Substitution mitCatridecacog das Risiko von Blutungen verringern.
Quelle:
EMA-Epar
Aclidiniumbromid von der EU-Kommission für COPD-Patienten zugelassen
Die EU-Kommission hat Aclidiniumbromid (Bretaris Genuair, Eklira Genuair) für die Behandlung von Patienten mit chronisch-obstruktiver Lungenerkrankung zugelassen.
Aclidiniumbromid (Bretaris Genuair, Eklira Genuair) wird als bronchodilatatorische Dauertherapie bei Erwachsenen mit chronisch-obstruktiver Lungenerkrankung (COPD) angewendet, um deren Symptome zu lindern.

Aclidiniumbromid ist ein kompetitiver, selektiver Muskarin-Rezeptor-Antagonist (Anticholinergikum) mit einer längeren Bindungsdauer an die M3-Rezeptoren als die M2-Rezeptoren. M3-Rezeptoren regeln die Kontraktion der glatten Muskulatur der Luftwege. Inhaliertes Aclidiniumbromid wirkt lokal in den Lungen, wo es an die M3-Rezeptoren der glatten Bronchialmuskulatur antagonistisch bindet und eine Bronchodilatation bewirkt.
Die Rate an systemischen anticholinergen Nebenwirkungen ist gering, da Aclidiniumbromid schnell im Plasma abgebaut wird.
Quelle:
EMA-Epar
Aclidiniumbromid (Bretaris Genuair, Eklira Genuair) wird als bronchodilatatorische Dauertherapie bei Erwachsenen mit chronisch-obstruktiver Lungenerkrankung (COPD) angewendet, um deren Symptome zu lindern.
Aclidiniumbromid ist ein kompetitiver, selektiver Muskarin-Rezeptor-Antagonist (Anticholinergikum) mit einer längeren Bindungsdauer an die M3-Rezeptoren als die M2-Rezeptoren. M3-Rezeptoren regeln die Kontraktion der glatten Muskulatur der Luftwege. Inhaliertes Aclidiniumbromid wirkt lokal in den Lungen, wo es an die M3-Rezeptoren der glatten Bronchialmuskulatur antagonistisch bindet und eine Bronchodilatation bewirkt.
Die Rate an systemischen anticholinergen Nebenwirkungen ist gering, da Aclidiniumbromid schnell im Plasma abgebaut wird.
Quelle:
EMA-Epar
Perampanel von der EU-Kommission bei Epilepsie zugelassen
Die EU-Kommission hat Perampanel (Fycompa, Eisa) als Zusatztherapie fokaler Anfälle mit oder ohne sekundäre Generalisierung bei Epilepsiepatienten ab 12 Jahren zugelassen.

Perampanel (Fycompa, Eisa) ist ein nicht kompetitiver Antagonist am ionotropen α-Amino-3-hydroxy-5-methyl-4-isoxazolproprionsäure (AMPA)-Glutamat-Rezeptor auf post-synaptischen Neuronen.
Der genaue Mechanismus, über den Perampanel seine antiepileptischen Wirkungen vermittelt, ist noch nicht bekannt.
Die Wirksamkeit wurde unter anderem in drei Plazebo-kontrollierten Studien mit Patienten ab 12 Jahren nachgewiesen. Häufigste unerwünschte Wirkungen waren Benommenheit und Somnolenz.
Quelle:
Teduglutid von der EU-Kommission für Patienten mit Kurzdarmsyndrom zugelassen
Die EU-Kommission hat Teduglutid (Revestive, Takeda) für die Behandlung von Patienten mit Kurzdarmsyndrom zugelassen.
Teduglutid (Revestive, Takeda) ist zur Behandlung von Erwachsenen mit Kurzdarmsyndrom indiziert. Nach einem chirurgischen Eingriff sollte zunächst eine Phase der intestinalen Adaption abgewartet werden, und die Patienten sollten sich in einer stabilen Phase befinden.
Teduglutid ist ein rekombinant hergestelltes Analogon des Glucacon-like Peptids 2 (GLP-2), das an der Regeneration des Darmepithels beteiligt sein soll. Teduglutid ist ein Peptid mit einer Länge von 33 Aminosäuren, bei dem das Alanin an der 2. Position des N-Terminus durch ein Glycin ersetzt wurde.

Im Vergleich zum natürlichen GLP-2 resultiert der Austausch dieser einzelnen Aminosäure in einer In-vivo-Resistenz gegen den Abbau durch die Dipeptidylpeptidase-IV (DPP-IV) und somit in einer verlängerten Halbwertszeit von Teduglutid.
In einigen präklinischen Studien zeigte sich, dass Teduglutid die Unversehrtheit der Schleimhaut erhält, indem es die Wiederherstellung des normalen Wachstums fördert. Dem liegt eine Zunahme der Darmzottenhöhe und der Darmkryptentiefe zugrunde.
Für die Behandlung des Kurzdarmsyndroms stehen derzeit keine Medikamente zur Verfügung. Teduglutid kann den Bedarf der Patienten an parenteraler Ernährung verringern. So konnte in der zulassungsrelevanten Studie die Menge an benötigter parenteraler Ernährung signifikant um 20 bis 100 % nach 20 und 24 Wochen im Vergleich zu Placebo reduziert werden. Nach 24 Wochen benötigten Patienten, die mit Teduglutid behandelt wurden, wöchentlich 4,4 Liter weniger. In der Kontrollgruppe reduzierte sich der Bedarf von parenteraler Ernährung auf 2,3 Liter, zuvor hatten die Patienten in der Verumgruppe 12,9 Liter und in der Kontrollgruppe 13,2 Liter wöchentlich erhalten. Die häufigsten Nebenwirkungen sind Bauchschmerzen und Blähungen, Infektionen der Atemwege, Übelkeit, Erbrechen und Reaktionen an der Injektionsstelle.
Quelle:
EPAR der EMA
Teduglutid (Revestive, Takeda) ist zur Behandlung von Erwachsenen mit Kurzdarmsyndrom indiziert. Nach einem chirurgischen Eingriff sollte zunächst eine Phase der intestinalen Adaption abgewartet werden, und die Patienten sollten sich in einer stabilen Phase befinden.
Teduglutid ist ein rekombinant hergestelltes Analogon des Glucacon-like Peptids 2 (GLP-2), das an der Regeneration des Darmepithels beteiligt sein soll. Teduglutid ist ein Peptid mit einer Länge von 33 Aminosäuren, bei dem das Alanin an der 2. Position des N-Terminus durch ein Glycin ersetzt wurde.
Im Vergleich zum natürlichen GLP-2 resultiert der Austausch dieser einzelnen Aminosäure in einer In-vivo-Resistenz gegen den Abbau durch die Dipeptidylpeptidase-IV (DPP-IV) und somit in einer verlängerten Halbwertszeit von Teduglutid.
In einigen präklinischen Studien zeigte sich, dass Teduglutid die Unversehrtheit der Schleimhaut erhält, indem es die Wiederherstellung des normalen Wachstums fördert. Dem liegt eine Zunahme der Darmzottenhöhe und der Darmkryptentiefe zugrunde.
Für die Behandlung des Kurzdarmsyndroms stehen derzeit keine Medikamente zur Verfügung. Teduglutid kann den Bedarf der Patienten an parenteraler Ernährung verringern. So konnte in der zulassungsrelevanten Studie die Menge an benötigter parenteraler Ernährung signifikant um 20 bis 100 % nach 20 und 24 Wochen im Vergleich zu Placebo reduziert werden. Nach 24 Wochen benötigten Patienten, die mit Teduglutid behandelt wurden, wöchentlich 4,4 Liter weniger. In der Kontrollgruppe reduzierte sich der Bedarf von parenteraler Ernährung auf 2,3 Liter, zuvor hatten die Patienten in der Verumgruppe 12,9 Liter und in der Kontrollgruppe 13,2 Liter wöchentlich erhalten. Die häufigsten Nebenwirkungen sind Bauchschmerzen und Blähungen, Infektionen der Atemwege, Übelkeit, Erbrechen und Reaktionen an der Injektionsstelle.
Quelle:
EPAR der EMA
Montag, 24. September 2012
Dronedaron: EMA empfiehlt neue Kontraindikation
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, als neue Kontraindikation bei der Behandlung mit Dronedaron (Multaq, Sanofi) die gleichzeitige Behandlung mit Dabigatran aufzunehmen.
Quelle:
Mitteilung der EMA vom 20. September 2012
Quelle:
Mitteilung der EMA vom 20. September 2012
Vildagliptin: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung von Vildagliptin (Galvus, Jalra, Xiliarx, Novartis) zu erweitern.
Vildagliptin (Galvus, Jalra, Xiliarx, Novartis) soll nun auch als Dreifachtherapie in Kombination mit einem Sulfonylharnstoff und Metformin sowie in Kombination mit Insulin mit oder ohne Metformin zur Behandlung von Patienten mit Diabetes mellitus Typ 2 eingesetzt werden können, deren Erkrankung mit Ernährung und/oder körperlicher Aktivität nicht ausreichend kontrolliert werden kann.
Quelle:
Mitteilung der EMA vom 20. September 2012
Vildagliptin (Galvus, Jalra, Xiliarx, Novartis) soll nun auch als Dreifachtherapie in Kombination mit einem Sulfonylharnstoff und Metformin sowie in Kombination mit Insulin mit oder ohne Metformin zur Behandlung von Patienten mit Diabetes mellitus Typ 2 eingesetzt werden können, deren Erkrankung mit Ernährung und/oder körperlicher Aktivität nicht ausreichend kontrolliert werden kann.
Quelle:
Mitteilung der EMA vom 20. September 2012
Everolimus: EMA empfiehlt Zulassungserweiterung bei tuberöser Sklerose
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung von Everolimus (Votubia, Novartis) für die Behandlung renaler Angiolipome bei Patienten mit tuberöser Sklerose zu erweitern.
Everolimus (Votubia, Novartis) ist ein m-Tor-Hemmer, der nun auch zur Therapie von erwachsenen Patienten mit tuberöser Sklerose (TSC) assoziiert mit renalen Angiomyolipomen eingesetzt werden soll.
Quelle:
Mitteilung der EMA vom 20. September 2012
Everolimus (Votubia, Novartis) ist ein m-Tor-Hemmer, der nun auch zur Therapie von erwachsenen Patienten mit tuberöser Sklerose (TSC) assoziiert mit renalen Angiomyolipomen eingesetzt werden soll.
Quelle:
Mitteilung der EMA vom 20. September 2012
Tenofovirdisoproxil: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, von Tenofovirdisoproxil (Viread, Gilead) eine neue Zubereitung sowie neue Stärken mit erweiterten Anwendungsgebieten zuzulassen.
Tenofovirdisoproxil als oral applizierbares Granulat (33 mg/g) soll in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 2 bis unter 6 Jahren zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen. Bei Kindern über 6 Jahren sowie bei Erwachsenen soll das Granulat eingesetzt werden können, wenn sie die Tabletten nicht einnehmen können.
Das Granulat soll zudem bei Erwachsenen zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn sie Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose
- dekompensierter Lebererkrankung
Das Granulat soll bei Jugendlichen von 12 bis 18 Jahren zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn sie Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose
Als neue Stärke sollen Filmtabletten mit 123, 163 und 204 mg zugelassen werden
Die Tabletten mit 123 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 17 und 22 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Tabletten mit 163 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 22 und 28 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Tabletten mit 204 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 22 und 35 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Zulassung für die 245-mg-Tabletten soll in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Jugendlichen im Alter von 12 bis unter 18 Jahren erweitert werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Zudem sollen sie bei Jugendlichen von 12 bis 18 Jahren zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn diese Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose.
Quelle:
Mitteilung der EMA vom 20. September 2012
Tenofovirdisoproxil als oral applizierbares Granulat (33 mg/g) soll in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 2 bis unter 6 Jahren zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen. Bei Kindern über 6 Jahren sowie bei Erwachsenen soll das Granulat eingesetzt werden können, wenn sie die Tabletten nicht einnehmen können.
Das Granulat soll zudem bei Erwachsenen zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn sie Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose
- dekompensierter Lebererkrankung
Das Granulat soll bei Jugendlichen von 12 bis 18 Jahren zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn sie Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose
Als neue Stärke sollen Filmtabletten mit 123, 163 und 204 mg zugelassen werden
Die Tabletten mit 123 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 17 und 22 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Tabletten mit 163 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 22 und 28 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Tabletten mit 204 mg sollten sie in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Kindern im Alter von 6 bis unter 12 Jahren mit einem Körpergewicht zwischen 22 und 35 kg zugelassen werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Die Zulassung für die 245-mg-Tabletten soll in Kombination mit anderen antiretroviral wirkenden Substanzen für die Behandlung von HIV-infizierten Jugendlichen im Alter von 12 bis unter 18 Jahren erweitert werden, die gegen NRTI resistent sind oder diese nicht vertragen.
Zudem sollen sie bei Jugendlichen von 12 bis 18 Jahren zur Behandlung der chronischen Hepatitis B eingesetzt werden können, wenn diese Tabletten nicht nehmen können, und zwar bei
- kompensierter Lebererkrankung mit nachgewiesener aktiver viraler Replikation, dauerhaft
erhöhten Alaninaminotransferase- (ALT-)Werten im Serum und histologischem Nachweis einer
aktiven Entzündung und/oder Fibrose.
Quelle:
Mitteilung der EMA vom 20. September 2012
Samstag, 22. September 2012
Linagliptin: EMA empfiehlt Zulassungserweiterung zur Kombination mit Insulin
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung von Linagliptin (Trajenta, Boehringer Ingelheim - in D nicht im Handel) zu erweitern.
Linagliptin (Trajenta, Boehringer Ingelheim) soll nun auch in Kombination mit Insulin mit oder ohne Metformin zur Behandlung von Patienten mit Diabetes mellitus Typ 2 eingesetzt werden können, deren Erkrankung mit Ernährung und/oder körperlicher Aktivität nicht ausreichend kontrolliert werden kann.
Quelle:
Mitteilung der EMA vom 20. September 2012
Linagliptin (Trajenta, Boehringer Ingelheim) soll nun auch in Kombination mit Insulin mit oder ohne Metformin zur Behandlung von Patienten mit Diabetes mellitus Typ 2 eingesetzt werden können, deren Erkrankung mit Ernährung und/oder körperlicher Aktivität nicht ausreichend kontrolliert werden kann.
Quelle:
Mitteilung der EMA vom 20. September 2012
Tadalafil: EMA empfiehlt Zulassungserweiterung
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung des Phosphodiesterasehemmers Tadalafil (Cialis, Janssen-Cilag) zu erweitern.
Tadalafil (Cialis, Janssen-Cilag) soll nun auch zur Behandlung der Symptome einer benignen Prostatahyperplasie einschließlich der erektilen Dysfunktion eingesetzt werden können.
Quelle:
Mitteilung der EMA vom 20. September 2012
Tadalafil (Cialis, Janssen-Cilag) soll nun auch zur Behandlung der Symptome einer benignen Prostatahyperplasie einschließlich der erektilen Dysfunktion eingesetzt werden können.
Quelle:
Mitteilung der EMA vom 20. September 2012
Bevacizumab: EMA empfiehlt Zulassungsweiterung für Ovarialkarzinom
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung von Bevacizumab (Avastin, Roche) zur Behandlung von Frauen mit rezidiviertem platinsensitivem Ovarialkarzinom in Kombination mit Carboplatin und Gemcitabin zu erweitern, wenn sie bisher noch nicht mit einem Angiogenesehemmer behandelt worden sind.
Quelle:
Mitteilung der EMA vom 20. September 2012
Quelle:
Mitteilung der EMA vom 20. September 2012
Apixaban: EMA empfiehlt Zulassungserweiterung für Patienten mit Vorhhofflimmern
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, die Zulassung von Apixaban (Eliquis, BristolMyersSquibb, Pfizer) für die Prävention von Schlaganfall und Embolien bei Patienten mit Vorhofflimmern zu erweitern.
Apixaban (Eliquis, BristolMyersSquibb, Pfizer) ist ein oraler Faktor-Xa-Hemmer, der nun auch zur Prävention von Schlaganfall und systemischen Embolien bei Erwachsenen mit Vorhhofflimmern eingesetzt werden soll, die mindestens einen Risikofaktor aufweisen wie Schlaganfall oder TIA in der Anamnese, Alter ab 75 Jahren, Hypertonie, Diabetes mellitus, Herzinsuffizienz ab NYHA-Klasse II.
Außerdem sollen in die Fachinformation zwei weitere Kontraindikationen aufgenommen werden.
Quelle:
Mitteilung der EMA vom 20. September 2012
Apixaban (Eliquis, BristolMyersSquibb, Pfizer) ist ein oraler Faktor-Xa-Hemmer, der nun auch zur Prävention von Schlaganfall und systemischen Embolien bei Erwachsenen mit Vorhhofflimmern eingesetzt werden soll, die mindestens einen Risikofaktor aufweisen wie Schlaganfall oder TIA in der Anamnese, Alter ab 75 Jahren, Hypertonie, Diabetes mellitus, Herzinsuffizienz ab NYHA-Klasse II.
Außerdem sollen in die Fachinformation zwei weitere Kontraindikationen aufgenommen werden.
Quelle:
Mitteilung der EMA vom 20. September 2012
Ingenolmebutat: EMA empfiehlt Zulassung für aktinische Keratose
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, Ingenolmebutat (Picato, Leo Pharma) zur topischen Therapie von aktinischer Keratose bei Erwachsenen zuzulassen.
Ingenolmebutat (Picato, Leo Pharma) ist ein Diterpen, das im Milchsaft der Wolfsmilch (Euphorbia peplus) vorkommt. Nach Ergebnissen präklinischer Studien wirkt es als pleiotropes Agens, das einen raschen Zelltod induziert und Immunreaktionen auslöst, die über die spezifische Aktivierung von Proteinkinase C delta vermittelt werden. Es soll nun als Gel für die lokale Behandlung von nicht-hyperkeratotischen, nicht-hypertrophen aktinischen Keratosen bei Erwachsenen eingesetzt werden können.
Wirksamkeit und Verträglichkeit wurden in vier multizentrischen, randomisierten, doppelblinden Studien im Vergleich zur Anwendung des Vehikels bei Patienten mit aktinischer Keratose untersucht. Das Gel wurde über zwei bis drei Tage angewendet. Der primäre Endpunkt, die Beseitigung der Hautveränderung nach 57 Tagen, wurde mit Ingenolmebutat bei signifikant mehr Patienten erreicht, als in den Vergleichsgruppen.
Quellen:
Mitteilung der EMA vom 20. September 2012
Lebwohl M, et al. NEJM 2012;366:1010-9.
Ingenolmebutat (Picato, Leo Pharma) ist ein Diterpen, das im Milchsaft der Wolfsmilch (Euphorbia peplus) vorkommt. Nach Ergebnissen präklinischer Studien wirkt es als pleiotropes Agens, das einen raschen Zelltod induziert und Immunreaktionen auslöst, die über die spezifische Aktivierung von Proteinkinase C delta vermittelt werden. Es soll nun als Gel für die lokale Behandlung von nicht-hyperkeratotischen, nicht-hypertrophen aktinischen Keratosen bei Erwachsenen eingesetzt werden können.
Wirksamkeit und Verträglichkeit wurden in vier multizentrischen, randomisierten, doppelblinden Studien im Vergleich zur Anwendung des Vehikels bei Patienten mit aktinischer Keratose untersucht. Das Gel wurde über zwei bis drei Tage angewendet. Der primäre Endpunkt, die Beseitigung der Hautveränderung nach 57 Tagen, wurde mit Ingenolmebutat bei signifikant mehr Patienten erreicht, als in den Vergleichsgruppen.
Quellen:
Mitteilung der EMA vom 20. September 2012
Lebwohl M, et al. NEJM 2012;366:1010-9.
Aflibercept: EMA empfiehlt Zulassung für Makuladegeneration
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, den Angiogenesehemmer Aflibercept (Eylea, Bayer AG) für die Behandlung der feuchten Makuladegeneration zuzulassen.
Aflibercept (Eylea, Bayer AG) ist ein Angiogenesehemmer, der den Vascular endothelial growth factor-A hemmt. Er soll für die Behandlung von Erwachsenen mit feuchter altersbedingter Makuladegeneration eingesetzt werden können. Die Anwendung sollte nur durch Ärzte erfolgen, die mit intravitrealen Injektionen Erfahrung haben.
Quelle:
Mitteilung der EMA vom 20. September 2012
Aflibercept (Eylea, Bayer AG) ist ein Angiogenesehemmer, der den Vascular endothelial growth factor-A hemmt. Er soll für die Behandlung von Erwachsenen mit feuchter altersbedingter Makuladegeneration eingesetzt werden können. Die Anwendung sollte nur durch Ärzte erfolgen, die mit intravitrealen Injektionen Erfahrung haben.
Quelle:
Mitteilung der EMA vom 20. September 2012
Linaclotid von der EMA zur Zulassung empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 die Zulassung von Linaclotid (Constella, Almirall) zur Behandlung des Reizdarmsyndroms mit Verstopfung empfohlen.
Linaclotid (Constella, Almirall) ist ein oral applizierbares Peptid aus 14 Aminosäuren, das als Agonist des Guanylat-C-Rezeptors fungiert, der auf der lumenseitigen Darmoberfläche vorkommt.

Linaclotid wird kaum resorbiert, es wirkt nach Einnahme direkt an den lumenseitigen Rezeptoren. Die Erhöhung der intrazellulären cGMP-Konzentration stimuliert die Sekretion von Chlorid und Hydrogencarbonat in das Darmlumen, vor allem durch Aktivierung des CFTR-Ionenkanals (Cystic fibrosis transmembrane conductance regulator). Dies führt im Darm zu einem erhöhten Flüssigkeitsvolumen und einer Beschleunigung des Transits. In Tiermodellen beschleunigte Linaclotid den gastrointestinalen Transit und verringerte intestinale Schmerzen. In klinischen Studien veränderte Linaclotid die Stuhlkonsistenz und erhöhte die Stuhlfrequenz.
Die FDA hat Linaclotid Ende August 2012 zugelassen (siehe med | pharm | text -Blog).
Quelle:
Pressemitteilung der EMA vom 21. September 2012
Linaclotid (Constella, Almirall) ist ein oral applizierbares Peptid aus 14 Aminosäuren, das als Agonist des Guanylat-C-Rezeptors fungiert, der auf der lumenseitigen Darmoberfläche vorkommt.

Linaclotid wird kaum resorbiert, es wirkt nach Einnahme direkt an den lumenseitigen Rezeptoren. Die Erhöhung der intrazellulären cGMP-Konzentration stimuliert die Sekretion von Chlorid und Hydrogencarbonat in das Darmlumen, vor allem durch Aktivierung des CFTR-Ionenkanals (Cystic fibrosis transmembrane conductance regulator). Dies führt im Darm zu einem erhöhten Flüssigkeitsvolumen und einer Beschleunigung des Transits. In Tiermodellen beschleunigte Linaclotid den gastrointestinalen Transit und verringerte intestinale Schmerzen. In klinischen Studien veränderte Linaclotid die Stuhlkonsistenz und erhöhte die Stuhlfrequenz.
Die FDA hat Linaclotid Ende August 2012 zugelassen (siehe med | pharm | text -Blog).
Quelle:
Pressemitteilung der EMA vom 21. September 2012
Ananasenzyme von der EMA zur Zulassung empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Zulassungsbehörde (EMA) hat in seiner Sitzung vom September 2012 empfohlen, ein Konzentrat proteolytischer Enzyme aus Bromelain (NexoBrid, MediWound) für die Zulassung zur Wundbehandlung zu empfehlen.
NexoBrid (MediWound) hat seit 2002 den Orphan-Drug-Status. Es enthält eine Mischung proteolytischer Enzyme aus Bromelain, das aus Stamm und Früchten der Ananaspflanze gewonnen wird. Es soll nun lokal zur Ablösung von fest haftendem Wundschorf bei schweren Verbrennungen eingesetzt werden. Nach Ergebnissen klinischer Studien verkürzt das Enzympräparat die Zeit bis zur Entfernung von Wundschorf und verringert den Bedarf an chirurgischem Debridement.
Bei nicht sachrgerechter Anwendung von NexoBrid können sich jedoch die Heilungszeiten verlängern und das Risiko von Wundinfektionen erhöhen. Daher soll der Hersteller verpflichtet werden, ein Trainingprogramm und entsprechende Fortbildungsmaterialien für die Anwender zur Verfügung zu stellen.
Quelle:
Pressemitteilung der EMA vom 21. September 2012
NexoBrid (MediWound) hat seit 2002 den Orphan-Drug-Status. Es enthält eine Mischung proteolytischer Enzyme aus Bromelain, das aus Stamm und Früchten der Ananaspflanze gewonnen wird. Es soll nun lokal zur Ablösung von fest haftendem Wundschorf bei schweren Verbrennungen eingesetzt werden. Nach Ergebnissen klinischer Studien verkürzt das Enzympräparat die Zeit bis zur Entfernung von Wundschorf und verringert den Bedarf an chirurgischem Debridement.
Bei nicht sachrgerechter Anwendung von NexoBrid können sich jedoch die Heilungszeiten verlängern und das Risiko von Wundinfektionen erhöhen. Daher soll der Hersteller verpflichtet werden, ein Trainingprogramm und entsprechende Fortbildungsmaterialien für die Anwender zur Verfügung zu stellen.
Quelle:
Pressemitteilung der EMA vom 21. September 2012
Pramipexol: FDA prüft Risiko einer Herzinsuffizienz
Die Food and Drug Administration (FDA) prüft, ob der Dopaminagonist Pramipexol das Risiko einer Herzinsuffizienz erhöht.
Die FDA geht Hinweisen aus klinischen und epidemiologischen Studien nach, die allerdings methodische Schwächen aufweisen. Die Behörde sieht in den Daten bislang keinen Beweis für ein erhöhtes Risiko einer kardialen Schädigung durch den Dopaminagonisten, will jedoch weitere Untersuchungen veranlassen.
Quelle:
FDA Sicherheitsmitteilung vom 19. September 2012
Die FDA geht Hinweisen aus klinischen und epidemiologischen Studien nach, die allerdings methodische Schwächen aufweisen. Die Behörde sieht in den Daten bislang keinen Beweis für ein erhöhtes Risiko einer kardialen Schädigung durch den Dopaminagonisten, will jedoch weitere Untersuchungen veranlassen.
Quelle:
FDA Sicherheitsmitteilung vom 19. September 2012
Dienstag, 18. September 2012
Bulletin zur Arzneimittelsicherheit erschienen
Das "Bulletin zur Arzneimittelsicherheit – Informationen aus BfArM und PEI" erscheint viermal im Jahr und informiert aus beiden Bundesoberbehörden über aktuelle Aspekte der Risikobewertung von Arzneimitteln.
Themen der Ausgabe vom September 2012 sind u. a.:
Brivudin und 5-Fluoropyrimidine – eine potenziell tödliche Interaktion
Qualitätsmangel beim Notfallarzneimittel Anapen® – ein Rückruf bis zur Patientenebene
Wirksamkeit und Risiken von Paracetamol
Falldefinition für die progressive multifokale Leukenzephalopathie (PML) nach Therapie mit monoklonalen Antikörpern
Daten zur Pharmakovigilanz von Impfstoffen aus dem Jahr 2010
Bedeutung von nichtinterventionellen Unbedenklichkeitsstudien im Rahmen von Post-Authorisation Safety Studies (PASS) – Aspekte der neuen Pharmakovigilanzgesetzgebung
Wie lassen sich Anti-Drug-Antikörper verhindern? – EU-Forschungskonsortium mit Beteiligung des PEI.
Quelle:
AkdÄ - News vom 14.9.2012
Sonntag, 16. September 2012
Teriflunomid von der FDA für die Behandlung der MS zugelassen
Die Food and Drug Administration (FDA) hat Teriflunomid (Aubagio, Sanofi Aventis) für die Behandlung von Patienten mit schubförmig remittierender MS zugelassen.
Teriflunomid (Aubagio, Sanofi Aventis) ist der aktive Metabolit von Leflunomid, das als Arava® für die Behandlung der rheumatoiden Arthritis zugelassen ist. Es hemmt die Dihydroorotat-Dehydrogenase (DHODH) in den Mitochondrien, die ein Schlüsselenzym bei der Pyrimidin-Synthese ist. Dadurch hemmt es die Proliferation und Funktion von aktivierten Lymphozyten, nicht jedoch von ruhenden Zellen. Darüber hinaus wurden weitere Mechanismen beschrieben wie eine gestörte Signalübertragung im Nuclear factor κB (NF-κB)-Signalweg. Verschiedene Daten weisen darauf hin, dass Teriflunomid ein Immunmodulator ist, der diverse Manifestationen von Autoimmunerkrankungen unterdrücken kann, ohne die Infektionsabwehr zu beeinträchtigen.

Teriflunomid wird einmal täglich oral eingenommen, es ist für die Behandlung von Patienten mit schubförmig-remittierender MS zugelassen. In den klinischen Studien war die Schubrate unter Teriflunomid etwa 30 % geringer als in den Plazebo-Gruppen.
Häufigste Nebenwirkungen in den Studien waren Diarrhö, erhöhte Leberwerte, Übelkeit und Haarverlust. Vor Therapiebeginn und während der Behandlung sollte die Leberfunktion der Patienten überprüft werden.
Bei Frauen im gebärfähigen Alter muss vor Therapiebeginn ein negativer Schwangerschaftstest vorliegen, außerdem müssen sie empfängnisverhütende Maßnahmen durchführen.
Quelle:
Presseinformation der Food and Drug Administration (FDA) vom 12. September 2012
Teriflunomid (Aubagio, Sanofi Aventis) ist der aktive Metabolit von Leflunomid, das als Arava® für die Behandlung der rheumatoiden Arthritis zugelassen ist. Es hemmt die Dihydroorotat-Dehydrogenase (DHODH) in den Mitochondrien, die ein Schlüsselenzym bei der Pyrimidin-Synthese ist. Dadurch hemmt es die Proliferation und Funktion von aktivierten Lymphozyten, nicht jedoch von ruhenden Zellen. Darüber hinaus wurden weitere Mechanismen beschrieben wie eine gestörte Signalübertragung im Nuclear factor κB (NF-κB)-Signalweg. Verschiedene Daten weisen darauf hin, dass Teriflunomid ein Immunmodulator ist, der diverse Manifestationen von Autoimmunerkrankungen unterdrücken kann, ohne die Infektionsabwehr zu beeinträchtigen.

Teriflunomid wird einmal täglich oral eingenommen, es ist für die Behandlung von Patienten mit schubförmig-remittierender MS zugelassen. In den klinischen Studien war die Schubrate unter Teriflunomid etwa 30 % geringer als in den Plazebo-Gruppen.
Häufigste Nebenwirkungen in den Studien waren Diarrhö, erhöhte Leberwerte, Übelkeit und Haarverlust. Vor Therapiebeginn und während der Behandlung sollte die Leberfunktion der Patienten überprüft werden.
Bei Frauen im gebärfähigen Alter muss vor Therapiebeginn ein negativer Schwangerschaftstest vorliegen, außerdem müssen sie empfängnisverhütende Maßnahmen durchführen.
Quelle:
Presseinformation der Food and Drug Administration (FDA) vom 12. September 2012
Mittwoch, 5. September 2012
Bosutinib von der FDA zugelassen
Die Food and Drug Administration (FDA) hat Bosutinib (Bosulif, Pfizer) als Orphan Drug für die Behandlung von Patienten mit chronischer myeloischer Leukämie (CML) zugelassen.
Bosutinib (Bosulif, Pfizer) ist ein Tyrosinkinase-Inhibitor, der für die Behandlung von Patienten mit Philadelphia-Chromosom-positiver CML, akzelerierter CML und CML in der Blastenphase eingesetzt werden kann, die auf andere Therapien resistent sind oder diese nicht vertragen.

Wirksamkeit und Verträglichkeit von Bosutinib wurden in einer Studie mit 546 Erwachsenen mit chronischer, akzelerierte oder Blasten-CML untersucht. Bei allen Patienten war die Erkrankung nach Therapie mit Imatinib oder Imatinib gefolgt von Dasatinib und/oder Nilotinib fortgeschritten, oder sie konnten eine dieser Therapie nicht vertragen. Alle Patienten wurden mit Bosutinib behandelt.
Bei Patienten mit chronischer CML wurde die Wirksamheit anhand der Major cytogenetic response (MCyR) in den ersten 24 Behandlungswochen bestimmt. 34 % der Patienten, die zuvor mit Imatinib behandelt worden waren, erreichten eine MCyR nach 24 Wochen. Bei 52,8 % der Patienten, die zu einem beliebigen Zeitpunkt eine MCyR erreichten, hielt sie mindestens 18 Monate an. 27 % der Patienten, die zuvor mit Imatinib gefolgt von Dasatinib und/oder Nilotinib behandelt worden waren, erreichten eine MCyR in den ersten 24 Wochen. Das Ansprechen hielt bei 51,4 % aller Responder mindestens 9 Monate an.
Bei 33 % der Patienten mit akzelerierter CML und bei 15 % mit Blasten-CML und Vorbehandlung mit Imatinib kam es zu einem kompletten hämatologischen Ansprechen.
Häufigste Nebenwirkungen waren Durchfall, Übelkeit, Thrombozytopenie, Erbrechen, Bauchschmerzen, Hautausschlag, Anämie, Fieber und Fatigue.
Quelle:
Pressemitteilung der FDA vom 4. September 2012
Bosutinib (Bosulif, Pfizer) ist ein Tyrosinkinase-Inhibitor, der für die Behandlung von Patienten mit Philadelphia-Chromosom-positiver CML, akzelerierter CML und CML in der Blastenphase eingesetzt werden kann, die auf andere Therapien resistent sind oder diese nicht vertragen.
Wirksamkeit und Verträglichkeit von Bosutinib wurden in einer Studie mit 546 Erwachsenen mit chronischer, akzelerierte oder Blasten-CML untersucht. Bei allen Patienten war die Erkrankung nach Therapie mit Imatinib oder Imatinib gefolgt von Dasatinib und/oder Nilotinib fortgeschritten, oder sie konnten eine dieser Therapie nicht vertragen. Alle Patienten wurden mit Bosutinib behandelt.
Bei Patienten mit chronischer CML wurde die Wirksamheit anhand der Major cytogenetic response (MCyR) in den ersten 24 Behandlungswochen bestimmt. 34 % der Patienten, die zuvor mit Imatinib behandelt worden waren, erreichten eine MCyR nach 24 Wochen. Bei 52,8 % der Patienten, die zu einem beliebigen Zeitpunkt eine MCyR erreichten, hielt sie mindestens 18 Monate an. 27 % der Patienten, die zuvor mit Imatinib gefolgt von Dasatinib und/oder Nilotinib behandelt worden waren, erreichten eine MCyR in den ersten 24 Wochen. Das Ansprechen hielt bei 51,4 % aller Responder mindestens 9 Monate an.
Bei 33 % der Patienten mit akzelerierter CML und bei 15 % mit Blasten-CML und Vorbehandlung mit Imatinib kam es zu einem kompletten hämatologischen Ansprechen.
Häufigste Nebenwirkungen waren Durchfall, Übelkeit, Thrombozytopenie, Erbrechen, Bauchschmerzen, Hautausschlag, Anämie, Fieber und Fatigue.
Quelle:
Pressemitteilung der FDA vom 4. September 2012
Telavancin: EMA widerruft Zulassung
Die Europäische Zulassungsbehörde (EMA) hat die Zulassung für das Antibiotikum Telavancin (Vibativ) widerrufen.
Quelle:
Human Medicines Highlights der EMA vom August 2012
Quelle:
Human Medicines Highlights der EMA vom August 2012
Axitinib von der EU-Kommision zugelassen
Die EU-Kommission hat Axitinib (Inlyta, Pfizer) für die Behandlung von erwachsenen Patienten mit fortgeschrittenem Nierenzellkarzinom nach Versagen einer vorangegangenen Therapie mit Sunitinib oder einem Zytokin zugelassen.
Axitinib (Inlyta, Pfizer) ist ein VEGF-Hemmer, der die über VEGF vermittelte Proliferation von Endothelzellen und deren Überleben inhibiert. Axitinib ist nun für die Behandlung von Erwachsenen mit fortgeschrittenem Nierenzellkarzinom (RCC) zugelassen, die zuvor mit Sunitinib oder einem Zytokin behandelt worden sind oder nicht auf diese Therapie angesprochen haben.

Die Zulassung basiert vorwiegend auf einer internationalen, randomisierten, offenen Studie bei 723 Patienten mit fortgeschrittenem RCC nach Versagen einer vorherigen systemischen Therapie. Primärer Endpunkt der Studie war das progressionsfreie Überleben (PFS). 361 Patienten erhielten Axitinib 5 mg oral zweimal täglich, 362 Patients wurden mit Sorafenib 400 mg oral zweimal täglich bis zur Progression der Erkrankung oder inakzeptablen Toxizität behandelt. Alle Patienten hatten einen ECOG-Performancestatus von 0 oder 1 und waren zuvor mit Sunitinib, Temsirolimus, Bevacizumab oder Zytokinen behandelt worden. Ausgeschlossen waren Patienten mit unkontrolliertem Bluthochdruck. Das PFS war bei den Axitinib-Patienten signifikant besser als bei den Sorafenib-Patienten (HR=0,67; 95% KI: 0,54-0,81; p< 0,0001, Log-rank.Test). Das mediane PFS unter Axitinib lag bei 6,7 Monaten (95%-KI: 6,3 -8,6), unter Sorafenib bei 4,7 Monaten (95%-KI: 4,6-5,6). Häufigte (≥ 20 %) Nebenwirkungen unter Axitinib waren Diarrhö Hypertonie, Fatigue, verminderter Appetit, Übelkeit, Dysphonie, Hand-Fuß-Syndrom, Gewichtsabnahme, Erbrechen, Asthenie und Verstopfung. Weitere schwere Nebenwirkungen bei den Axitinib-Patienten waren hypertensive Krise, arterielle und venöse Thrombosen, Blutungen, gastrointestinale Perforation und Fistelbildung und reversibles Leukenzephalopathie-Syndrom.
Dienstag, 4. September 2012
Daunorubicin: Lieferengpass angekündigt
Die Firma Pfizer hat angekündigt an, dass sie Anfang
September einen vorübergehenden Lieferengpass bei Daunoblastin
®
erwartet.
Daunorubicin ist ein seit 40 Jahren angewendetes, hochwirksames und nicht verzichtbares Medikament für die Behandlung von Patienten mit akuter Leukämie bei Erwachsenen und Kindern. Die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) und die DGHO Deutsche Gesellschaft für Hämatologie und Onkologie e.V. sehen diese Entwicklung mit großer Besorgnis.
Quelle:
Brivudin: Rote-Hand-Brief zu Wechselwirkungen mit 5-Fluoropyrimidinen
Der Hersteller von Brivudin (Zostex) hat einen Rote-Hand-Brief zu potenziell tödlichen Wechselwirkungen zwischen Brivudin und 5-Fluoropyrimidinen verschickt.
Die AkdÄ hat 2006 in einer Bekanntgabe im Deutschen Ärzteblatt auf potenziell tödlich verlaufende Wechselwirkungen zwischen Brivudin (Zostex®) und 5-Fluoropyrimidinen hingewiesen. Brivudin hemmt irreversibel die Dihydropyrimidin-Dehydrogenase (DPD). Dieses Enzym reguliert den Metabolismus von natürlichen Nukleosiden und von Pyrimidin-Derivaten wie 5-FU. Die Hemmung der DPD führt zur Akkumulation und verstärkten Toxizität von 5-FU.
Der Hersteller von Zostex® informiert jetzt in einem Rote-Hand-Brief über die wichtige Kontraindikation und Vorsichtsmaßnahmen: Brivudin darf nicht zusammen mit 5-Fluoropyrimidin-haltigen Arzneimitteln angewendet werden. Hierzu gehören z. B. 5-FU, dessen Prodrugs Capecitabin, Floxuridin und Tegafur sowie Flucytosin einschließlich topischer Zubereitungen oder Kombinationsprodukten mit diesen Wirkstoffen. Eine Therapie mit 5-Fluoropyrimidinen darf frühestens vier Wochen nach der letzten Anwendung von Brivudin begonnen werden (zusätzliche Vorsichtsmaßnahme: Bestimmung der DPD-Emzymaktivität). Bei einer versehentlichen Gabe von 5-Fluoropyrimidinen an mit Brivudin behandelte Patienten müssen beide Arzneimittel sofort abgesetzt und Maßnahmen zur Reduzierung der 5-Fluoropyrimidin-Toxizität ergriffen werden: stationäre Aufnahme, Verhütung von systemischen Infektionen und einer Dehydratation, Gabe von Uridin.
Quelle:
AkdÄ Drug Safety Mail vom 4. September 2012
Die AkdÄ hat 2006 in einer Bekanntgabe im Deutschen Ärzteblatt auf potenziell tödlich verlaufende Wechselwirkungen zwischen Brivudin (Zostex®) und 5-Fluoropyrimidinen hingewiesen. Brivudin hemmt irreversibel die Dihydropyrimidin-Dehydrogenase (DPD). Dieses Enzym reguliert den Metabolismus von natürlichen Nukleosiden und von Pyrimidin-Derivaten wie 5-FU. Die Hemmung der DPD führt zur Akkumulation und verstärkten Toxizität von 5-FU.
Der Hersteller von Zostex® informiert jetzt in einem Rote-Hand-Brief über die wichtige Kontraindikation und Vorsichtsmaßnahmen: Brivudin darf nicht zusammen mit 5-Fluoropyrimidin-haltigen Arzneimitteln angewendet werden. Hierzu gehören z. B. 5-FU, dessen Prodrugs Capecitabin, Floxuridin und Tegafur sowie Flucytosin einschließlich topischer Zubereitungen oder Kombinationsprodukten mit diesen Wirkstoffen. Eine Therapie mit 5-Fluoropyrimidinen darf frühestens vier Wochen nach der letzten Anwendung von Brivudin begonnen werden (zusätzliche Vorsichtsmaßnahme: Bestimmung der DPD-Emzymaktivität). Bei einer versehentlichen Gabe von 5-Fluoropyrimidinen an mit Brivudin behandelte Patienten müssen beide Arzneimittel sofort abgesetzt und Maßnahmen zur Reduzierung der 5-Fluoropyrimidin-Toxizität ergriffen werden: stationäre Aufnahme, Verhütung von systemischen Infektionen und einer Dehydratation, Gabe von Uridin.
Quelle:
AkdÄ Drug Safety Mail vom 4. September 2012
Denosumab: Rote-Hand-Brief wegen Hypokalzämie mit Todesfolge
Der Hersteller von Denosumab (Xgeva) informiert in einem Rote-Hand-Brief über schwere Fälle von Hypokalzämie mit teilweise tödlichem Ausgang und gibt Empfehlungen zur Minimierung des Risikos.
Eine Hypokalzämie kann zu jedem Zeitpunkt während der Behandlung mit Denosumab auftreten. Anzeichen und Symptome umfassen eine Veränderung des mentalen Status, Tetanie, Krampfanfälle sowie eine Verlängerung des QTc-Intervalls. Vor Behandlung mit Denosumab muss eine bereits bestehende Hypokalzämie korrigiert werden. Alle Patienten müssen ergänzend Kalzium und Vitamin D erhalten – außer bei bestehender Hyperkalzämie. Bei Auftreten einer Hypokalzämie ist möglicherweise eine zusätzliche Kalziumgabe erforderlich. Bei schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) oder bei dialysepflichtigen Patienten besteht ein höheres Risiko, eine Hypokalzämie zu entwickeln.
Quelle:
AkDÄ Drug Safety Mail vom 4. September 2012
Eine Hypokalzämie kann zu jedem Zeitpunkt während der Behandlung mit Denosumab auftreten. Anzeichen und Symptome umfassen eine Veränderung des mentalen Status, Tetanie, Krampfanfälle sowie eine Verlängerung des QTc-Intervalls. Vor Behandlung mit Denosumab muss eine bereits bestehende Hypokalzämie korrigiert werden. Alle Patienten müssen ergänzend Kalzium und Vitamin D erhalten – außer bei bestehender Hyperkalzämie. Bei Auftreten einer Hypokalzämie ist möglicherweise eine zusätzliche Kalziumgabe erforderlich. Bei schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) oder bei dialysepflichtigen Patienten besteht ein höheres Risiko, eine Hypokalzämie zu entwickeln.
Quelle:
AkDÄ Drug Safety Mail vom 4. September 2012
Levofloxacin: Rote-Hand-Brief wegen schwerwiegenden Nebenwirkungen
Der Hersteller von Levofloxacin (Tavanic) hat einen Rote-Hand-Brief zu Indikationseinschränkungen, neuen schwerwiegenden Nebenwirkungen und Vorsichtsmaßnahmen bei der Anwendung von Levofloxacin Filmtabletten und Infusionslösung verschickt.
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittel-Agentur (EMA) kam nach Abschluss eines Verfahrens zur Harmonisierung der nationalen Produktinformationen zu dem Ergebnis, dass bestimmte Anwendungsgebiete von Levofloxacin spezifiziert bzw. eingeschränkt werden müssen. Nach Auswertung sicherheitsrelevanter Daten wurden darüber hinaus die Aufnahme neuer schwerwiegender Nebenwirkungen einschließlich entsprechender Warnhinweise beschlossen und die Produkt- und Fachinformationen aktualisiert.
Einschränkung folgender Indikationen von Levofloxacin (sollte nur angewendet werden, wenn Antibiotika,
die üblicherweise zur Initialbehandlung eingesetzt werden, nicht indiziert sind):
- akute bakterielle Sinusitis,
- akute Exazerbation einer chronischen Bronchitis,
- ambulant erworbene Pneumonie,
- komplizierte Haut- und Weichteilinfektionen.
• Neue schwerwiegende Nebenwirkungen:
- hypoglykämisches Koma,
- ventrikuläre Arrhythmie und Torsade de pointes, ventrikuläre Tachykardie, die zum Herzstillstand
führen kann,
- tödliche Fälle von akutem Leberversagen,
- benigne intrakranielle Hypertonie,
- vorübergehender Sehverlust,
- Pankreatitis,
- Verschlimmerung der Symptome einer bestehenden Myasthenia gravis,
- Bänder- und Muskelrisse,
- Hörverlust.
• Ergänzende Warnhinweise zur Überwachung von Patienten und zur Vermeidung oder Minderung von
einigen zuvor genannten Nebenwirkungen.
Quelle:
AkdÄ Drug Safety Mail vom 3. September 2012
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittel-Agentur (EMA) kam nach Abschluss eines Verfahrens zur Harmonisierung der nationalen Produktinformationen zu dem Ergebnis, dass bestimmte Anwendungsgebiete von Levofloxacin spezifiziert bzw. eingeschränkt werden müssen. Nach Auswertung sicherheitsrelevanter Daten wurden darüber hinaus die Aufnahme neuer schwerwiegender Nebenwirkungen einschließlich entsprechender Warnhinweise beschlossen und die Produkt- und Fachinformationen aktualisiert.
Einschränkung folgender Indikationen von Levofloxacin (sollte nur angewendet werden, wenn Antibiotika,
die üblicherweise zur Initialbehandlung eingesetzt werden, nicht indiziert sind):
- akute bakterielle Sinusitis,
- akute Exazerbation einer chronischen Bronchitis,
- ambulant erworbene Pneumonie,
- komplizierte Haut- und Weichteilinfektionen.
• Neue schwerwiegende Nebenwirkungen:
- hypoglykämisches Koma,
- ventrikuläre Arrhythmie und Torsade de pointes, ventrikuläre Tachykardie, die zum Herzstillstand
führen kann,
- tödliche Fälle von akutem Leberversagen,
- benigne intrakranielle Hypertonie,
- vorübergehender Sehverlust,
- Pankreatitis,
- Verschlimmerung der Symptome einer bestehenden Myasthenia gravis,
- Bänder- und Muskelrisse,
- Hörverlust.
• Ergänzende Warnhinweise zur Überwachung von Patienten und zur Vermeidung oder Minderung von
einigen zuvor genannten Nebenwirkungen.
Quelle:
AkdÄ Drug Safety Mail vom 3. September 2012
Domperidon: ventrikuläre Arrhythmien und plötzlicher Herztod
Neuere Studien zeigen, dass die Einnahme des Prokinetikums Domperidon (Motilium) mit einem gering (etwa 1,5-fach) erhöhten Risiko für schwerwiegende ventrikuläre Herzrhythmusstörungen und plötzlichen Herztod assoziiert ist.
Domperidon sollte in der niedrigsten wirksamen Dosis (möglichst nicht über 30 mg täglich) und nicht gleichzeitig mit Medikamenten verordnet werden, die ebenfalls zu einer QT-Verlängerung führen. Besondere Indikationsstellung ist erforderlich bei Patienten mit vorbestehender Verlängerungen der QT-Zeit, bei Elektrolytstörungen, bei vorbestehenden Herzerkrankungen oder Einnahme von kardialen Medikamenten, bei Patienten älter als 60 Jahre und bei der Notwendigkeit der Verordnung von mehr als 30 mg Domperidon pro Tag.
Quelle:
UAW-News International, AkdÄ Drug Safety Mail vom 3. September 2012
Domperidon sollte in der niedrigsten wirksamen Dosis (möglichst nicht über 30 mg täglich) und nicht gleichzeitig mit Medikamenten verordnet werden, die ebenfalls zu einer QT-Verlängerung führen. Besondere Indikationsstellung ist erforderlich bei Patienten mit vorbestehender Verlängerungen der QT-Zeit, bei Elektrolytstörungen, bei vorbestehenden Herzerkrankungen oder Einnahme von kardialen Medikamenten, bei Patienten älter als 60 Jahre und bei der Notwendigkeit der Verordnung von mehr als 30 mg Domperidon pro Tag.
Quelle:
UAW-News International, AkdÄ Drug Safety Mail vom 3. September 2012
Sonntag, 2. September 2012
Ceftarolinfosamil von der Europäischen Kommission zugelassen
Die EU-Kommission hat Ceftarolinfosamil (Zinforo, AstraZeneca) für die Behandlung von Patienten mit komplizierten Haut- und Weichgewebeinfektionen und ambulant erworbenen Atemwegsinfektionen zugelassen.
Ceftarolinfosamil (Zinforo, AstraZeneca) ist ein Cephalosporin das durch Plasma-Phosphatasen schnell in die Wirkform Ceftarolin überführt wird.
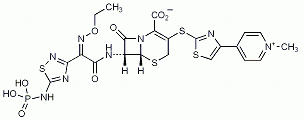
Es bindet an PBP2a und ist damit gegen MRSA und Methicillin-resistente Staphylococcus epidermidis (MRSE) sowie Penicillin-resistente Streptokokken und Vancomycin-resistente Enterokokken wirksam. Das breite Aktivitätsspektrum beinhaltet auch gramnegative Erreger, aber keine ESBL-bildenden Enterobacteriaceae und nur bedingt Anaerobier. Häufigste Nebenwirkungen in den Studien waren ein positiver Coombs-Test, Hautauschlag, Pruritus, Kopfschmerzen, Benommenheit, Phlebitis, Diarrhö, Nausea, Erbrechen, Bauchschmerzen, erhöhte Transaminasen, Pyrexie und Reaktionen an der Infusionsstelle.
Quelle:
Biospace vom 28. August 2012
Ceftarolinfosamil (Zinforo, AstraZeneca) ist ein Cephalosporin das durch Plasma-Phosphatasen schnell in die Wirkform Ceftarolin überführt wird.
Es bindet an PBP2a und ist damit gegen MRSA und Methicillin-resistente Staphylococcus epidermidis (MRSE) sowie Penicillin-resistente Streptokokken und Vancomycin-resistente Enterokokken wirksam. Das breite Aktivitätsspektrum beinhaltet auch gramnegative Erreger, aber keine ESBL-bildenden Enterobacteriaceae und nur bedingt Anaerobier. Häufigste Nebenwirkungen in den Studien waren ein positiver Coombs-Test, Hautauschlag, Pruritus, Kopfschmerzen, Benommenheit, Phlebitis, Diarrhö, Nausea, Erbrechen, Bauchschmerzen, erhöhte Transaminasen, Pyrexie und Reaktionen an der Infusionsstelle.
Biospace vom 28. August 2012
Ruxolitinib von der EU-Kommission zugelassen
Die EU-Kommission hat den JAK-Hemmer Ruxolitinib (Jakavi, Novartis) für die Behandlung von Patienten mit Myelofibrose zugelassen.
Ruxolitinib (Jakavi, Novartis) ist ein Proteinkinase-Inhibitor (JAK-Hemmer), der Janus-assoziierte Kinasen (JAK) hemmt. JAKs sind an der Signalübertragung von Zytokinen und Wachstumsfaktoren beteiligt, die für die Hämatopoese und Immunfunktionen wichtig sind.

Ruxolitinib ist zugelassen zur Behandlung von Erwachsenen mit primärer Myelofibrose (chronisch idiopathischer Myelofibrose), Post-Polycythaemia-vera-Myelofibrose oder Post-essentieller-Thrombozythämie-Myelofibrose.
Es führt zu einer Verkleinerung der Milz und wirkt auf weitere Symptome der Myelofibrose. Häufigste Nebenwirkungen sind Thrombozytopenie, Anämie und Blutungen.
In den USA ist Ruxolitinib als Jakafi seit November 2011 für diese Indikation zugelassen.
Quelle
Biospace vom 28. August 2012
Ruxolitinib (Jakavi, Novartis) ist ein Proteinkinase-Inhibitor (JAK-Hemmer), der Janus-assoziierte Kinasen (JAK) hemmt. JAKs sind an der Signalübertragung von Zytokinen und Wachstumsfaktoren beteiligt, die für die Hämatopoese und Immunfunktionen wichtig sind.
Ruxolitinib ist zugelassen zur Behandlung von Erwachsenen mit primärer Myelofibrose (chronisch idiopathischer Myelofibrose), Post-Polycythaemia-vera-Myelofibrose oder Post-essentieller-Thrombozythämie-Myelofibrose.
Es führt zu einer Verkleinerung der Milz und wirkt auf weitere Symptome der Myelofibrose. Häufigste Nebenwirkungen sind Thrombozytopenie, Anämie und Blutungen.
In den USA ist Ruxolitinib als Jakafi seit November 2011 für diese Indikation zugelassen.
Quelle
Biospace vom 28. August 2012
Enzalutamid von der FDA bei Prostatakarzinom zugelassen
Die Food and Drug Administration (FDA) hat Enzalutamid (Xtandi, Astellas) für die Behandlung von Männern mit Prostatakarzinom im fortgeschrittenen Stadium zugelassen.
Enzalutamid (Xtandi, Astellas) ist ein Androgenrezeptorantagonist, der mit einer etwa fünffach höheren Affinität an den Androgenrezeptor bindet als Bicalutamid.

Es wurde von der FDA beschleunigt zugelassen für die Behandlung von Männern mit metastasiertem, Kastrations-resistentem, progredientem oder rezidiviertem Prostatakarzinom, die mit Docetaxel vorbehandelt wurden.
Sicherheit und Wirksamkeit wurden in einer Studie mit 1.199 Patienten untersucht, die zuvor mit Docetaxel behandelt worden waren. Das mediane Gesamtüberleben der Patienten mit Enzalutamid betrug 18,4 Monate im Vergleich zu 13,6 Monaten der Patienten unter Plazebo.
Häufigste Nebenwirkungen waren Schwäche oder Fatigue, Rückenschmerzen, Diarrhö, Gelenkschmerzen, Hitzewallungen, Gelenkschwellungen, muskuloskeletale Schmerzen, Kopfschmerzen, Infektionen der oberen und unteren Atemwege, Benommenheit, Rückenmarkskompression und Cauda-equina-Syndrom, Schlafstörungen, Blut im Urin, Angst und Bluthochdruck.
Quelle:
Pressemitteilung der FDA vom 31. August 2012
Enzalutamid (Xtandi, Astellas) ist ein Androgenrezeptorantagonist, der mit einer etwa fünffach höheren Affinität an den Androgenrezeptor bindet als Bicalutamid.
Es wurde von der FDA beschleunigt zugelassen für die Behandlung von Männern mit metastasiertem, Kastrations-resistentem, progredientem oder rezidiviertem Prostatakarzinom, die mit Docetaxel vorbehandelt wurden.
Sicherheit und Wirksamkeit wurden in einer Studie mit 1.199 Patienten untersucht, die zuvor mit Docetaxel behandelt worden waren. Das mediane Gesamtüberleben der Patienten mit Enzalutamid betrug 18,4 Monate im Vergleich zu 13,6 Monaten der Patienten unter Plazebo.
Häufigste Nebenwirkungen waren Schwäche oder Fatigue, Rückenschmerzen, Diarrhö, Gelenkschmerzen, Hitzewallungen, Gelenkschwellungen, muskuloskeletale Schmerzen, Kopfschmerzen, Infektionen der oberen und unteren Atemwege, Benommenheit, Rückenmarkskompression und Cauda-equina-Syndrom, Schlafstörungen, Blut im Urin, Angst und Bluthochdruck.
Quelle:
Pressemitteilung der FDA vom 31. August 2012
Linaclotid von der FDA bei chronischer Obstipation zugelassen
Die Food and Drug Administration (FDA) hat Linaclotid (Linzess, Ironwood, Forest) für die Behandlung von Patienten mit chronischer idiopathischer Obstipation und mit Reizdarmsyndrom mit Obstipation zugelassen.
Linaclotid (Linzess, Ironwood, Forest) ist ein oral applizierbares Peptid aus 14 Aminosäuren, das als Agonist des Guanylat-C-Rezeptors fungiert, der auf der lumenseitigen Darmoberfläche vorkommt.

Linaclotid wird kaum resorbiert, es wirkt nach Einnahme direkt an den lumenseitigen Rezeptoren. Die Erhöhung der intrazellulären cGMP-Konzentration stimuliert die Sekretion von Chlorid und Hydrogencarbonat in das Darmlumen, vor allem durch Aktivierung des CFTR-Ionenkanals (Cystic fibrosis transmembrane conductance regulator). Dies führt im Darm zu einem erhöhten Flüssigkeitsvolumen und einer Beschleunigung des Transits. In Tiermodellen beschleunigte Linaclotid den gastrointestinalen Transit und verringerte intestinale Schmerzen. In klinischen Studien veränderte Linaclotid die Stuhlkonsistenz und erhöhte die Stuhlfrequenz.
Quellen:
Pressemitteilung der FDA vom 30. August 2012
Amerikanische Gebrauchsinformation von Linzess
Linaclotid (Linzess, Ironwood, Forest) ist ein oral applizierbares Peptid aus 14 Aminosäuren, das als Agonist des Guanylat-C-Rezeptors fungiert, der auf der lumenseitigen Darmoberfläche vorkommt.

Quellen:
Pressemitteilung der FDA vom 30. August 2012
Amerikanische Gebrauchsinformation von Linzess
Sildenafil: FDA warnt vor Einsatz bei Kindern
Die Food and Drug Administration (FDA) warnt vor dem Einsatz von Sildenafil (Revatio) zur Behandlung der pulmonalen Hypertonie (PAH) bei Kindern. Der PDE-Hemmer ist nur bei Erwachsenen zugelassen.
Sildenafil (Revatio, Pfizer) ist für die Behandlung von Kindern mit PAH nicht zugelassen und von der Offl-Label-Anwendung rät die FDA aufgrund klinischer Daten ab, nach denen bei Einnahme hoher Dosen durch Kinder das Mortalitätsrisiko im Vergleich zur Anwendung niedriger Dosen erhöht wird.
Quelle:
FDA Drug Safety Communication vom 30. August 2012
Sildenafil (Revatio, Pfizer) ist für die Behandlung von Kindern mit PAH nicht zugelassen und von der Offl-Label-Anwendung rät die FDA aufgrund klinischer Daten ab, nach denen bei Einnahme hoher Dosen durch Kinder das Mortalitätsrisiko im Vergleich zur Anwendung niedriger Dosen erhöht wird.
Quelle:
FDA Drug Safety Communication vom 30. August 2012
Abonnieren
Kommentare (Atom)


